
INTRODUCTION
Over the past decade, artificial intelligence (AI) and machine learning (ML, a subfield of AI) tools have revolutionized various domains, from creating smart enhanced content in education 1 to strengthening e-mail security by improved spam detection 2 and even helping farmers optimize their irrigation methods. 3
In the healthcare sector, AI has made substantial contributions throughout academia and industry. Applications range from drug discovery and development 4, 5 to clinical trial optimization 6, 7 and drug manufacturing. 8 In 2021, the Food and Drug Administration (FDA) reported over 100 submissions of drug and biological applications using AI and ML components. 9 In 2023, we saw the first fully AI-generated drug reaching phase II clinical trials in humans, 10 potentially leading the way to a whole new generation of drugs.
While AI can help speed up the drug development pipeline in many ways, bringing new drugs to the market remains costly, time-consuming, and is prone to a high failure rate. 11, 12 Repurposing existing drugs for new therapeutic indications aims to reduce the cost and time of development, additionally seeing a much lower rate of clinical trial failure, especially at the early stages of the clinical study (while phase III of clinical trials remains essentially the same in terms of time and costs). Drug repurposing thus has the potential to dramatically decrease the time safe therapies may take to reach patients in need of a treatment. 13
At the level of accurate compound or target selection, drug repurposing is empowered both by the continuous development and improvement of high-throughput multi-omics technologies and the quick advances in computational methods. 14 More recently, AI/ML applications have played a crucial role in the way drugs can be repurposed. AI-based tools leveraging deep learning and neural networks are particularly promising to, for instance, screen compounds, predict new drug–target combinations, and assess adverse drug reactions, which can ultimately speed up the selection of existing targets and drugs for new applications. 15– 17
Although AI-based methods offer new and exciting opportunities in the field of drug repurposing, and drug development in general, they come with new and unique challenges.
In this review, we explore different AI/ML-based methods and their approach to complex problems related to drug repurposing, and we also highlight current obstacles, challenges, and perspectives within this field.
MACHINE LEARNING-BASED METHODS TO REPURPOSE DRUGS
Ziaurrehman Tanoli
Background and Significance of Machine Learning in Drug Repurposing
Drug repurposing can be executed through either target-based or phenotypic-based methodologies. Nonetheless, both approaches necessitate experimental validation in clinical settings, incurring substantial financial investments.
ChEMBL V-33 18 encompasses bioactivity data for 3492 approved small molecules, elucidating their interactions with specific targets. The Open Targets platform 19 stands out as one of the most comprehensive resources, cataloging associations between genes and over 21,000 diseases and symptoms. Given the vast number of potential drug–disease pairings—potentially reaching millions—conducting experimental tests on animals or in clinical trials becomes a formidable and costly challenge. 20 Consequently, the application of ML-based prediction methods offers an alternative, enabling the timely identification of prioritized drug–disease associations that can subsequently undergo validation in clinical trials.
Existing Tools and Machine Learning-Based Methods for Drug Repurposing
The drug discovery paradigm comprises several integral phases, including hit identification, assessment of drug safety, dose optimization, and drug repurposing, the latter focusing on the identification of new disease associations for already approved drugs. This discussion will concentrate specifically on tools and methodologies pertinent to drug repurposing.
In recent years, the field of drug repurposing has witnessed the development of numerous computational resources dedicated to supporting these endeavors. Notable examples include DrugRepo, 21 Drug Repurposing Hub, 22 repoDB, 23 and RepurposeDB, 24 along with an array of other web-based databases, collectively contributing directly or indirectly to the drug repurposing landscape. 25
In tandem with web-based resources, various prediction methods for drug repurposing have been introduced. PREDICT, 26 a widely utilized computational approach, leverages drug–drug and disease–disease networks, specifically focusing on 593 drugs and 313 diseases. Another innovative study presented a bipartite graph-based methodology aimed at uncovering novel drug indications by exploring their relationships with similar drugs. 27 The CMAF method employs matrix factorization to predict drug–disease associations based on the networks of drug and disease similarities. 28
It is crucial to note that these methods exhibit limitations, primarily in terms of coverage, as they are restricted to a limited set of drugs and disease indication pairs. Notably, their predictive capacity for disease associations related to drug combinations remains insufficient. Furthermore, the absence of interactive web applications for these methodologies restricts their practical utility and broader accessibility within the scientific community.
RepurposeDrugs: A Novel Machine Learning-Based Algorithm to Repurpose Drugs and Combinations for Hundreds of the Diseases
To overcome existing limitations and enhance drug repurposing capabilities for researchers lacking programming skills, we introduce RepurposeDrugs ( https://repurposedrugs.org/), an ML-based web portal offering a versatile approach to uncover novel relationships between drugs and diseases, encompassing both single and combination therapies. Our dataset for drug–disease associations underwent meticulous curation from the clinical trials database ( https://clinicaltrials.gov/). These associations were categorized as approved (positive class) or failed (negative class) based on the reported status of each clinical trial. Failed drug–indication pairs denote instances where at least two trials were terminated or withdrawn for a disease indication different from the drug’s original approval.
We fine-tuned two XGBoost-based regression models, one for single drugs and another for combinations. The positive dataset for the single-drug XGBoost model comprised 382 approved drugs and 190 diseases, while the negative dataset contained 409 drugs and 175 diseases. Similarly, for the drug combination model, the positive dataset included 65 approved combinations for 55 diseases, and the negative dataset comprised 62 drug combinations across 39 diseases.
These models were trained using eight distinct descriptor sets, encompassing two-dimensional (2D), three-dimensional (3D), and graph neural network (GNN)-based fingerprints of drugs, in addition to Lipinski’s rule of five (RO5) descriptors. Various descriptor types were incorporated to enhance the accuracy of the prediction model. The 2D fingerprints encoded structural information with descriptors such as ECFP4 (1024 bits) and ECFP6 (1024 bits), 29 MACCS (166 bits), 30 Klekota-Roth (4860 bits), 31 PubChem (881 bits), 32 and E-State (79 bits). 33 The 3D fingerprints (E3FP) provided spatial arrangement insights with a length of 4096 bits, 34 while GNN-based fingerprints (3DInfoMax) 35 captured molecular graph structures (256 bits). The fingerprint for each drug combination was determined through a logical OR operation on each bit of the individual drug fingerprints, effectively capturing features present in either drug of the combination.
Optimal XGBoost parameters were determined via Bayesian optimization and 10-fold cross-validation, minimizing overfitting while maximizing performance, with root mean square error (RMSE) as the primary performance metric. Note that cross-validation in this context refers to out-of-fold predictions, which are obtained by aggregating the predictions made on each validation set during the 10-fold cross-validation process. In cross-validation, the XGBoost prediction algorithm within RepurposeDrugs demonstrated a significant correlation of 0.75 for single drug–disease associations and 0.56 for drug combinations. Despite the lower correlation for drug combinations, attributed in part to the limited dataset size, future enhancements with additional trial data are anticipated to bolster the model’s predictive capabilities.
Moreover, RepurposeDrugs incorporates a conformal prediction module to filter out low-confidence predictions by setting a default confidence threshold of 0.8, focusing on the most promising drug–disease associations.
We have also validated RepurposeDrugs for predicting outcomes of phase III, phase II, and phase I trials. Information on these trials, which successfully completed various drug–disease associations, was extracted from https://clinicaltrials.gov/ For phase I compounds, RepurposeDrugs had a predicted approval likelihood of 29%, reflecting the exploratory nature of this phase. RepurposeDrugs’s predicted approval likelihood increased to 38% for phase II trials, aligning with the focus on efficacy. Most notably, phase III trials showed a significant increase to a 63% mean predicted likelihood, corresponding to their advanced stage and proximity to market approval. These results demonstrate our model’s capability to estimate the likelihood of drug approval at different stages of clinical trials.
Anticipated to be a valuable resource for the drug repurposing community, RepurposeDrugs is poised to facilitate innovative repurposing strategies and leverage existing data for predictive analysis. It could significantly reduce the costs associated with running clinical trials, by predicting the likelihood of drug approval for specific indications in advance, so that clinical trial priorities could be established.
DRUG TARGET PREDICTION AND REPOSITIONING USING MACHINE LEARNING INTEGRATION OF NETWORK-BASED, PATHWAY ENRICHMENT-BASED, AND DISEASE ENRICHMENT-BASED ANALYSES
Ezequiel Anokian
ML and AI methods have gained popularity for their ability to significantly expedite the drug development process and mitigate risks.
The Clarivate ML-driven pipeline for drug repurposing uses an input as simple as a disease term or disease-related genes, to detect novel connections between diseases and drugs. This largely automated system employs a compendium of algorithms: molecular network analysis, molecular pathway assessment, and disease similarity. The overall ML pipeline aims to assess whether a gene plays a role in the disease of interest, producing scores that are eventually integrated using ML (partial least square and recursive feature elimination 36 ) to generate a final ranking.
There is a wide diversity of ML-based approaches in drug repurposing based on how they work and the input data they require. 13, 37 For example, ligand similarity and molecular docking methods operate with target and ligand 3D structures to predict features such as binding affinity (BA) and stability. Other strategies for identifying suitable drug candidates rely on the “reverse transcriptional signature” principle, where compounds showcasing gene expression profiles opposite to that of the disease of interest (i.e. can revert expression profile back to normal levels) are prioritized.
The success of AI-based drug repurposing tools is subject to the quality and consistency of the data they utilize. Often, these models have been trained on data from public sources, such as DrugBank, PubChem, BindingDB, ICD9/10, Mesh, UniProtKB, and PDB. 38
Nevertheless, computational drug repurposing comes with significant challenges. For example, identification of suitable candidates demands a profound understanding of the molecular mechanisms underpinning diseases. Furthermore, the absence of a clear regulatory framework tailored to the distinctive challenges of drug repurposing has hampered its widespread adoption. 13
Among the main advantages, AI-based drug repurposing plays a significant role in the burgeoning field of personalized medicine, aiming to accommodate the diversity in disease manifestations, genetic variations, and treatment responses among patients. This not only expedites the development of targeted therapies but also empowers healthcare providers to select treatments with a higher probability of success, thereby minimizing trial-and-error approaches.
Computational drug repurposing also aligns with the growing awareness of the environmental impact of pharmaceutical production. Repurposing existing drugs diminishes the need for extensive resource-intensive manufacturing processes, contributing to a more environmentally responsible pharmaceutical industry.
Real-world data (RWD) have emerged as an alternative to traditional clinical trial data in recent years. 39 By encompassing this longitudinal information collected from electronic health records, insurance claims, and patient registries, RWD provide a comprehensive view of a drug’s performance in real-world scenarios. The integration of such large data into the model training and/or fine-tuning not only expedites the research process but also captures the nuances of patient heterogeneity and comorbidities often excluded from controlled clinical trials.
In conclusion, we are witnessing the ever-evolving field of AI-driven drug repurposing and how it is accelerating the overall drug discovery process at an unprecedented pace. The evolving landscape also demands new business models and a collaborative strategy involving academia, industry, and regulatory bodies to establish standardized protocols for evaluating repurposed drugs, ensuring both efficacy and safety.
By overcoming the obstacles, fostering collaboration, and adapting regulatory frameworks, AI-based drug repurposing could revolutionize the landscape of medicine, delivering timely and cost-effective solutions to pressing healthcare challenges.
ENHANCING DRUG REPURPOSING THROUGH GRAPH NEURAL NETWORKS AND LINK PREDICTION
Lucía Prieto Santamaría
The increasing availability of biomedical heterogeneous data obtained from improved multi-omics techniques has allowed science to redefine the way we conceive diseases, becoming more holistic and interrelated entities. 40 More importantly, it has opened up new horizons in the possible ways of searching for treatments. In this context, we find the process of drug repurposing, which aims to find new uses for already existing drugs.
There are multiple ways of advancing drug repurposing. Some of the most relevant strategies derive from computational approaches. 38 Particularly, representing this heterogeneous information in the form of graphs (also called networks) allows the exploitation and description of data structured in nodes connected by edges (also called links) in a very expressive manner (meaning they can include the implicit semantics underpinning a specific given structure). 41 In biomedical problems, nodes can represent diseases, symptoms, genes, proteins, genetic variants, non-coding RNAs, biological pathways, drugs, and so on, and the links may depict the connections between these different types of nodes. The advantages of arranging the information in this network structure are plenty, and its use toward tackling medicine challenges has been framed under the so-called network medicine. 42, 43
One of the major technical advancements over learning with graph structures lies in the new AI-based field of deep learning on graphs and its major formalism: GNNs. Contrasting with the process of developing sophisticated feature extractors (namely, feature engineering), deep learning addresses the learning problem by jointly learning representations of the raw input data and a predictive model for the task under study. This is usually approached by stacking multiple layers of differentiable non-linear transformations and training such model in an end-to-end fashion using gradient descent techniques. These resulting models are often called deep neural networks. Traditionally, deep learning has been employed to tackle Euclidean-shifted data problems. However, graph-framed data have an underlying structure that follows a non-Euclidean space. Extending deep neural models to these non-Euclidean domains, and specifically to graphs, 44 has been and is an emerging research area. 45, 46 The GNN 47, 48 formalism is a general framework for defining deep neural networks on graph data. The key idea is to generate representations (also called embeddings) for nodes or edges that depend on both the structure of the graph and the feature information that might be related to them.
Regarding drug repurposing, one of the most straightforward perspectives to use both network-structured information and GNNs is to target the link prediction task with regard to the disease–drug link type, as represented in Figure 1 . That is, using GNN pipelines to embed the information in the network (representing each node as a vector of features) to then decode these embedding vectors optimizing the prediction of new links of the type disease–drug. Following this idea, various models have been developed and presented previously. 15, 49, 50 In them, the integration of heterogeneous biomedical data organized as a graph has demonstrated its efficacy when combined with GNNs to address the drug repurposing challenge. This approach has also been employed to predict treatments for diseases with unclear mechanisms. 51
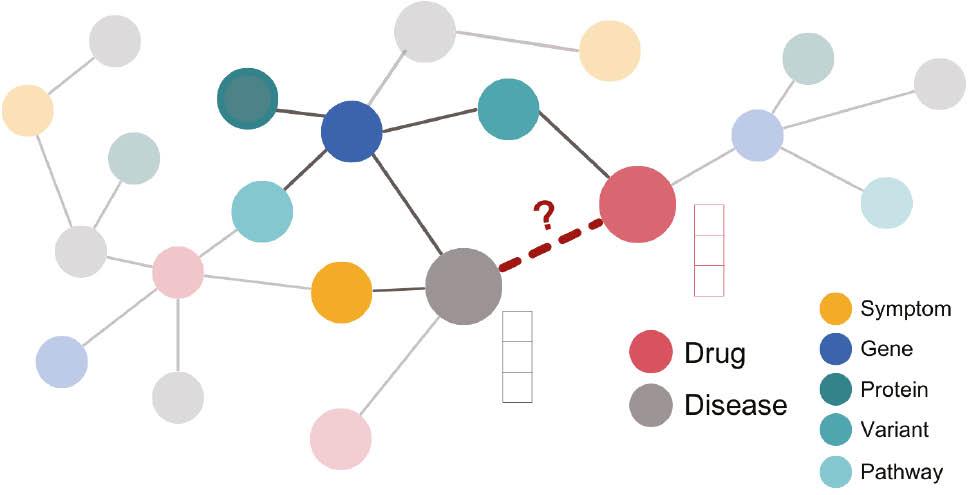
Representation of the disease–drug link prediction task in the context of drug repurposing. Each node depicts a biomedical entity: pink nodes represent drugs and gray nodes represent diseases. The rest of the node types include other relevant biomedical entities, such as symptoms and genes. The link prediction task in this case targets the disease–drug edge type. By means of graph neural networks (GNNs), the structural information of each node is first embedded in a vector representation, and then a decoding function prioritizes the potential disease–drug links.
Nonetheless, there are several limitations in the field and challenges to be addressed. The predictions made by GNN-based models, as any in silico repurposing model, need the input and interpretations of experts, as well as experimental confirmation. They act as a means of pinpointing repurposing opportunities that may warrant further examination. The complexity of biomedical data and the need to represent it in a meaningful way pose significant and opened research lines. Integrating diverse information sources and refining models to handle this complexity are ongoing tasks. Additionally, the need for large and diverse datasets for training robust models, as well as addressing issues related to data quality and bias when considering patient-centric information, have to be emphasized.
In general, the application of AI in drug repurposing offers a great potential to reshape traditional approaches, leading to faster identification of candidates for repurposing. Specifically, employing sophisticated GNN architectures to integrate both the biomedical network structure and the particular node features can be key to having multimodal systems that bring about better opportunities. In this sense, future research paths will involve refining these models to incorporate even more diverse and heterogeneous data sources. This will eventually enhance explainability and interpretability. In this direction, complementing AI-based pipelines with more classical data-driven methodologies (as the ones provided by network biology in general, and network medicine in particular) could lead to better and more understandable predictions.
DRUG RESPONSE PREDICTION WITH MACHINE LEARNING: A CRITICAL ASSESSMENT OF CURRENT APPROACHES
Judith Bernett, Markus List
Park divides computational repurposing into knowledge-based, phenotype-based, and signature-based approaches. 52 In knowledge-based approaches, information about drugs, their potential targets, and disease–gene or disease–protein associations are integrated into databases or knowledge graphs such as NeDRex, 53 from where new indications for existing drugs can be predicted. Phenotype-based repurposing aims to benefit from electronic health records through natural language processing. Finally, signature-based methods rely on available molecular profiling or OMICs data. Here, two diverse challenges can be addressed. First, OMICs data can help pinpoint disease mechanisms via implicating disease genes or proteins or, on a higher level, pathways and disease modules harboring potential drug targets. Another important direction is drug response prediction, an active research field fueled by the availability of large-scale drug response screens coupled with one or several layers of OMICs data. Such datasets represent a treasure trove for ML enthusiasts. They are easily accessible through projects such as Connectivity Map, 54 NCI-60, 55 GDSC, 56 or CCLE. 57 The goal here is not only to reveal drug–target relationships but to understand and predict how drug response depends on the cellular context.
Currently, the notion of cellular context is typically limited to cell lines and sometimes extends to patient-derived xenografts. 58 Still, the general principle can be extended to personalized medicine, where physicians may one day tailor treatments to individual patients to improve therapeutic outcomes. Due to easy access to these data, many ML methods have emerged for predicting drug responses. 59 However, this burgeoning field faces substantial challenges that remain inadequately addressed.
Most of the current methods engage in monotherapy drug response prediction (reviewed in detail in the work by Firoozbakht et al. 60 ), where the field follows the general trend where models become increasingly more complex. The hope is that deep learning strategies will learn latent data structures and understand complex non-linear associations that classical ML methods fail to identify. It should be noted though, that the datasets commonly used here have not grown much in recent years and often do not have the necessary size to train models with millions of parameters, making a breakthrough as in the case of AlphaFold 61 unlikely. Surprisingly, we still see that the prediction accuracy, typically measured through the correlation of predicted and observed drug response on a hold-out dataset, has continuously improved over recent years and has now reached almost optimal performance.
This leads to the impression that drug response prediction is a solved problem, but this is by far not the case. First, most published methods lack reproducible or even available implementations, rendering it difficult to objectively assess their performance. Second, existing methods rarely compare themselves against simpler baseline models, making the superiority of highly complex models questionable. Third, it can be shown that even a simple mean predictor, which just reports mean drug response in the training data, can achieve surprisingly good performance, questioning whether current methods actually learn cell-line-specific drug response as advertised and whether current performance measures are adequate. 62 Indeed, it can be shown that the current practice of reporting the correlation between predicted and observed drug response across all drugs is subject to Simpson’s paradox ( Figure 2 ) due to drug-specific average responses. Fourth, current data-splitting strategies allow models to overfit and to learn shortcuts. The standard practice of random splits, where drugs and cell lines can be found across training, test, and validation data, encourages data leakage. 63 Fifth, frequently used measures to quantify and summarize drug response, such as IC 50, EC 50, and Area Under the Curve (AUC) values, do not adequately capture the drug response dynamic. While the field has started to develop mitigation strategies such as the recently published CurveCurator, 64 existing methods do not generalize to unseen drug response data in the same cell lines, demonstrating that true generalization of findings across datasets and translation to patient care are far from reality.
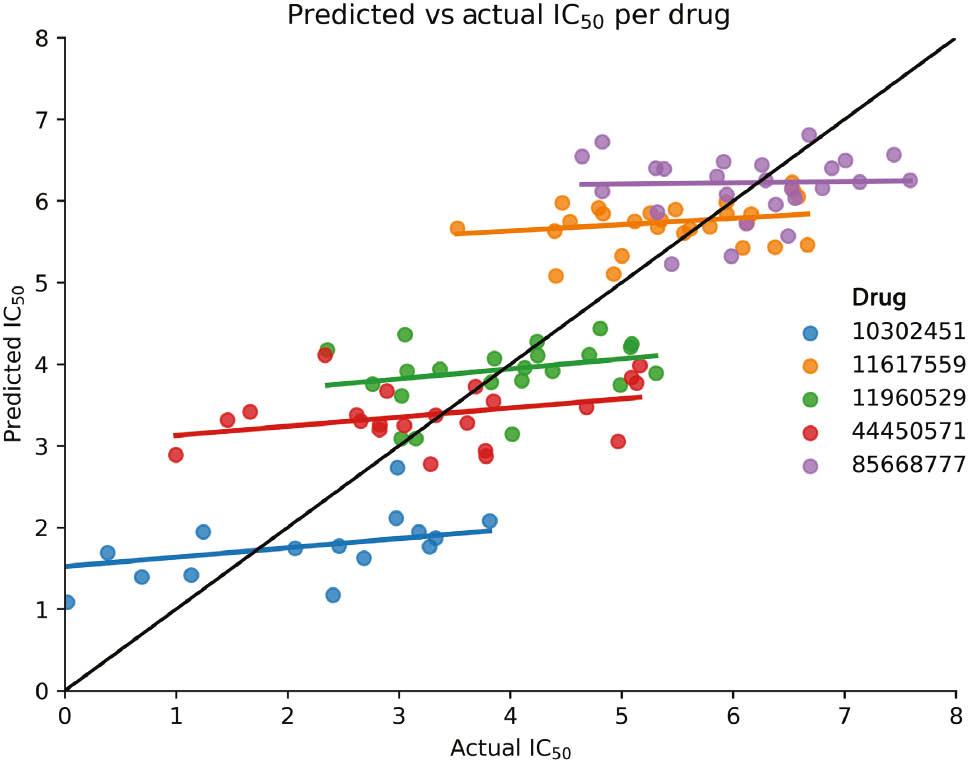
Visualization of the issue of evaluating the model performance across all drugs. While it seems like overall (black line), predicted IC 50s correlate almost perfectly with actual IC 50s, the evaluation per drug reveals that predictions concentrate mostly around the mean responses. The apparent correlation is removed (Simpson’s paradox).
Taken together, these issues create over-optimism about our current abilities to predict drug response and reveal a huge research gap that still needs to be closed before drug response prediction can become a routine aspect of patient care. Toward this goal, we advocate for better data standardization, more rigorous method evaluation, and benchmarking. Furthermore, most of the published drug response prediction methods rely exclusively on transcriptomics measurements, neglecting the multimodal nature of drug response. We envision that additional OMICs data, such as proteomics and phospho-proteomics data, could offer unique insights into the drug targets’ response and allow for building more robust methods. Furthermore, to equip deep learning models with the sample number necessary to learn complex associations, single-cell screening techniques such as Perturb-seq 65 are essential. Finally, future experimental and computational work should focus on predicting the response to drug combinations since this allows for synergistic effects at lower dosages and with fewer side effects. This is already the standard in cancer patient care but not reflected in computational modeling. A prerequisite for this is to improve the robustness and generalizability of existing approaches. These concerted efforts pave the way for more accurate and clinically relevant drug response predictions, ultimately enhancing patient care and treatment outcomes.
DATA-DRIVEN INDICATION DISCOVERY—EXPANDING THE POTENTIAL OF MEDICINES
Adrian Freeman
Within the pharmaceutical industry, we routinely divide ourselves into therapeutic areas and focus on projects that are related to a specific mechanism or target. As part of the drug discovery process a target is then used to drive the generation of a molecule, with many modalities now available as options to modulate the disease. This approach has not dramatically changed for several years and although the industry has become more efficient at driving molecules through the pipeline and success rates have increased, 66 there is still the requirement for delivering novel mechanisms of action in an expedited way for patient benefit.
The ever-increasing and rapid development of OMICS technologies is enabling data-driven-based approaches to be implemented within the pharma industry at multiple stages of the drug discovery pipeline to identify novel ways to progress ideas at multiple stages. The aim of our approach is to take a disease-agnostic view with a desire to remove the individual disease-centric view and start to consider mechanistic modules that form part of many diseases. This leads to considerations for gene/transcriptional network analysis across diseases, which may lead to combinatorial or temporal use of drugs within diseases.
An example of one approach that is being taken within different groups is the use of modified connectivity mapping approaches, 67, 68 where the transcriptomic signature of each compound is compared computationally with transcriptomic signatures of human diseases. As an example, differential gene expression signatures for compounds are generated by performing RNA sequencing on cell lines after exposure to two concentrations of each of the compounds. This analysis is completed in a blinded manner to the compound identity or chemistry and the generated genome-wide pattern of mRNA changes in cell-matched compound versus vehicle-treated samples. A disease transcriptomic library is then used at the differential gene expression data level. To identify novel clinical indications for each compound, a connectivity score is calculated by comparing each disease signature to each compound signature. The connectivity score aims to summarize the transcriptomic relationship between each compound and disease, such that a strongly negative score indicates that the compound will induce transcriptomic changes that may revert or “normalize” the disease signature.
The output from data-driven approaches are routinely employed and have had many successes within repurposing, but the landscape is now changing. Because of further enhancements with graph- and network-based approaches, the move from repositioning molecules to positioning using these methods is now more often used across the industrial and academic settings. Understanding the output from these models is crucial and validation is critical. There are core components that need to be considered e.g. data quality, code validation, and comprehensive metadata, among many others. The network analysis of many datasets has highlighted that the direct gene-to-protein relationship is not always the key driver of the disease phenotype and proven by lack of efficacy for certain medicines during clinical trials. The data-driven/network approaches are bringing forward the idea of interacting modules within the cell machinery, ranging from transcriptional regulation through to protein–protein interactions within the cytoplasm of cells. In addition, many of these modules interact and drive different phenotypes in different diseases. In the future, this should allow for therapies to be considered across diseases in an expedited manner.
CONCLUSION
In the field of drug discovery, AI- and ML-based methods became transformative tools for accelerating compound and drug target selection, particularly in the context of drug repurposing. Their impact is particularly relevant for precision medicine, where tailored treatments are essential, and for repurposing drugs to address rare diseases.
Network-structured information and GNNs provide a powerful framework for predicting new disease–drugs links, by integrating and organizing as a graph highly heterogeneous biomedical data and complex relationships.
Leveraging RWD allows for real-time assessment of a drug’s performance, which benefits patient stratification strategies, and improves patient recruitment and accurate monitoring. The increasing amount and quality of RWD can dramatically contribute to improving clinical trial success rates.
Incorporating additional layers of OMICs data, such as proteomics, phospho-proteomics, or single-cell technologies can offer the opportunity to build more comprehensive and robust predictive models.
Challenges persist in ensuring data coverage and quality, particularly needed for building and training high-quality and robust statistical models.
As the amount, complexity, and heterogeneity of input data keeps growing, there is a constant need to review and assess the operating procedures. Data security and protection also remain critical concerns.
While we increasingly rely on computational models to predict drug repurposing opportunities, there is constant need for input data selection and results interpretation by experts, combined with experimental validation, complementing the ever-increasing power of algorithms.
The rapid development of new methods underscores the importance of data quality, standardization, and rigorous and extensive benchmarking.
Despite the evolution of drug response prediction techniques, a gap persists for setting up standardized methods and accurate evaluation criteria.
Beyond data-centric challenges, ethical and regulatory considerations and challenges associated with AI- and ML-based tools remain critical and demand careful attention.
As this field evolves, we anticipate further breakthroughs and a broader scope of AI adoption in healthcare, constantly reshaping, rethinking, and reevaluating traditional approaches.