
INTRODUCTION
Floppy infant syndrome (FIS) is a recognizable neurological condition affecting infants in their early lives. It is characterized by generalized hypotonia donating a “rag doll” appearance, including the inability to maintain body posture, hanging the extremities on the sides, and lacking head control. Consequently, infants have poor motor skills, such as difficulties in feeding, speech, poor attention, and motivation, correlating with the disease severity. Myoferlin (MYOF) dysfunction is associated with muscular disorder ( Dong et al., 2019). Hypotonia is not a disorder per se but a manifestation of different neurological diseases, and the list of these diseases is exhaustive. However, it is generally categorized as those affecting the brain or the nerve/neuromuscular junction, with the central nervous system being the ultimate cause of FIS ( Igarashi, 2004).
MYOF belongs to the ferlin family along with FER-1, dysferlin ( DYSF), otoferlin, FER1L4, FER1L5 and FER1L6 ( Peulen et al., 2019). They share a similar structure, which includes several C2 domains and a carboxy-terminal transmembrane domain. They are fundamentally involved in cell processes requiring membrane fusion, such as endocytosis, exocytosis, and cell signaling ( Peulen et al., 2019).
MYOF is 180 kb long; it consists of 54 exons and encodes 9 splice variants, where 4 of these variants are not translated to protein ( Peulen et al., 2019). This gene is highly expressed in myoblasts and at the site of myoblasts fusing to myotubes, and therefore, it is essential for the fusion of myoblasts; it also plays a pivotal role in muscle development and in various muscle cell functions, including vesicle trafficking ( Demonbreun et al., 2010; Han et al., 2019). MYOF is also expressed in damaged myofibers and surrounding inflammatory cells, and mononuclear muscle, donating its function in muscle repair ( Demonbreun et al., 2010). MYOF null myoblasts in mice show a dystrophic muscle phenotype which is attributed to a defective myoblasts fusion ( Doherty et al., 2005). Furthermore, loss of MYOF in mice skeletal muscles leads to a reduced sensitivity in excitation–contraction coupling with deficient calcium release kinetics ( Barefield et al., 2021). In an adult female patient with cardiomyopathy and limb-girdle muscular dystrophy, a truncated variant of MYOF was identified ( Kiselev et al., 2019). Here, we present a case of an infant with generalized hypotonia with a novel MYOF gene mutation being the causative gene candidate for the current case.
METHODS
Ethical approval and sample collections
The study was approved by the local ethical committee of Center of Excellence in Genomic Medicine Research, King Abdulaziz University Jeddah, ethical approval number (013-CEGMR-02-ETH). A written informed consent for laboratory and genetic testing was obtained from the patient’s legal guardians. Sample collections and experimental work were done according to the international guidelines mentioned in the Declaration of Helsinki 2013. Blood samples from the patient were collected, and DNA was extracted from blood and stored in EDTA tubes as previously described ( Abdulkareem et al., 2019). The concentration of DNA was measured using NanodropTM 2000/2000c spectrophotometers.
Clinical history of the patient
This study included an 18-month-old male patient who presented with a lack of head support and an inability to sit or stand. He has a motor nerve condition with generalized hypotonia, which might be FIS. Till the age of 1 year and 6 months, the proband was unable to sit and stand and even unable to hold his head. His prenatal course was uncomplicated full term, and he was born at 39 weeks gestational age and 3.6 kg in weight. Developmental concerns were first noted at seventh month of age when he had developmental stagnation associated with poor feeding and failure to thrive. He was unable to sit unsupported and wheelchair dependent with head support. The detailed family pedigree was drawn after having information from the father as shown in Figure 1.

Whole exome sequencing
Whole exome sequencing (WES) technique was used to find the cause of the disease in the patient. DNA sample was used for WES in Illumina NextSeq 550 (High-Output v2 kit). The products were sequenced on an Illumina NextSeq instrument with 2 × 76 paired-end reads as previously described ( Naseer et al., 2020, 2021). The high quality was obtained by maintaining the quality control check on Illumina sequencing platforms. DNA library templates were constructed with accurate quantitation.
Obtained FASTQ files were converted to binary alignment map files, which were then converted to variant call format files. Further variants obtained were used for the identification of variants causing the disease based on homozygous or heterozygous state rare/novel (mutation annotation format + 0.01%) frequency, functional (predicted damage by polyphen/scale invariant feature transform), pathogenicity, genomic position, protein damaging effect, and linkage with the disease and its phenotype. Bioinformatics tools were used by applying various filters. The GRCh37 database was used for the alignment of the reference sequence. The obtained variants were further filtered to find out the disease linked with the identified variants in available public databases such as for allele frequencies <5.0% in the Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/), and nonsense, frameshift, and splice-site variants in disease associated genes with a minor allele frequency ≤1.0% were observed in gnomAD. The obtained list of variants was classified based on the American College of Medical Genetics (ACMG) and American College of Pathologists criteria and the identified variant lies in the criteria. ClinGen rule specifications ( https://www.clinicalgenome.org/working-groups/sequence-variant-interpretation/). Variants were reported according to human genome variation society nomenclature ( https://varnomen.hgvs.org/). We followed the standard guidelines of the ACMG. Deleterious effects and abnormalities were also identified using in-silico analysis for the structure and function of the identified variant leading to the disease. Mutation Taster ( http://www.mutationtaster.org/) was used for the identification of disease, further Exome Aggregation Consortium ( http://exac.broadinstitute.org/) was also used.
Sanger sequencing
After the results of WES, the identified variants was verified by using the Sanger sequencing technique in the remaining available family members. For polymerase chain reaction and sequencing the sets of the targeted primer were designed by using online primer 3 program and the designed primer sequences were as forward primer MYOF _3F:5-GCAGCCATACTACACACAGC-3′ and reverse primer MYOF_3R: 5′-CCAATGTCAGAGGCAGCAAA-3′. Sequencing data files were obtained from AB1 sequencing unit. The sequencing file was aligned with the reference sequence using the BioEdit software. The study was done under the guidelines of National Center for Biotechnology Information single-nucleotide polymorphism database.
RESULTS
Whole exome sequencing
WES results identified a homozygous abnormal splice variant in the Myoferlin gene where c.4982+1G>T p.Va11661fs changed as shown in Figure 2.
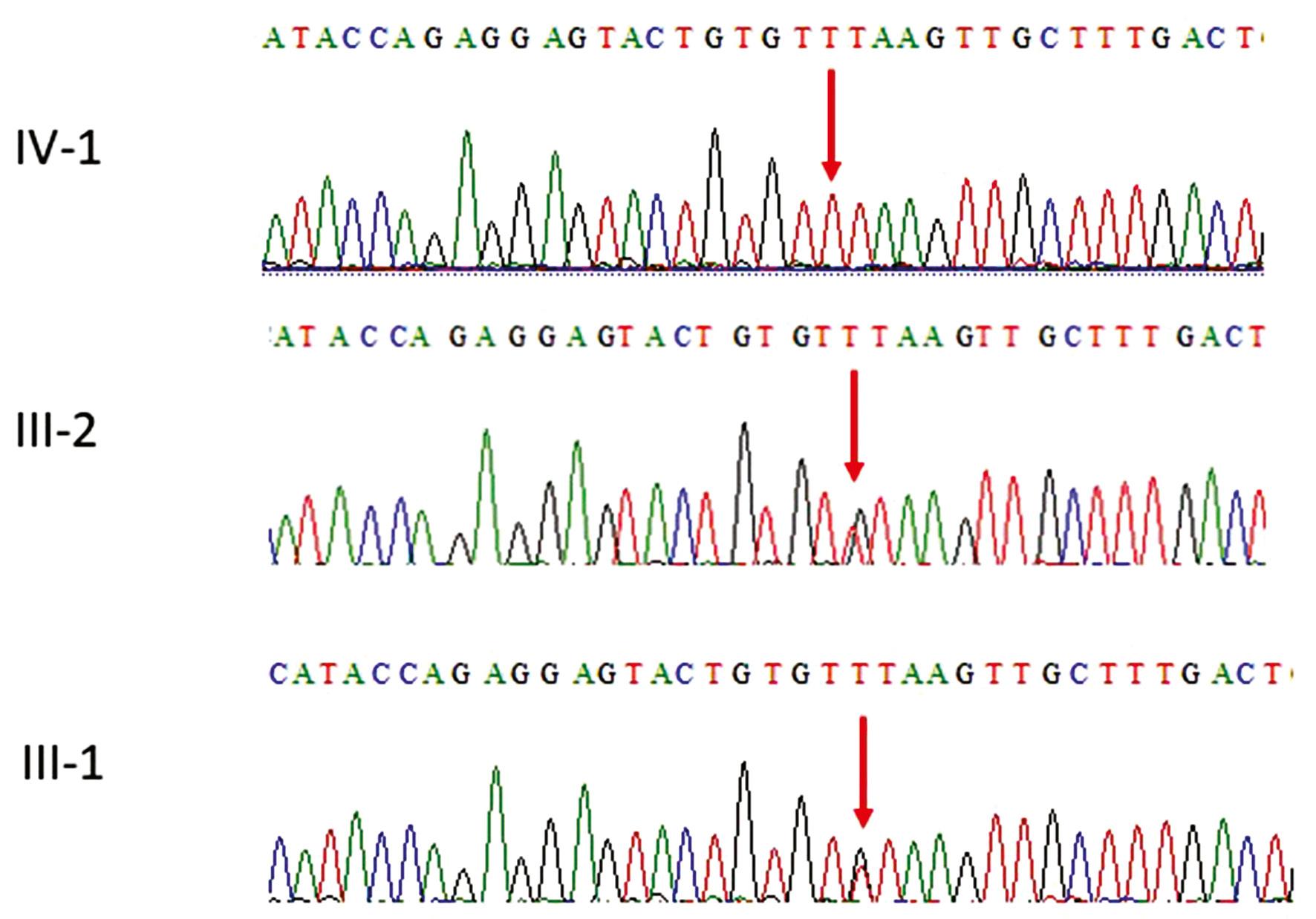
Representative electropherogram of the MYOF gene. Sanger sequencing results showing that the parents III-1 and III-2 are heterozygous carries having T/G on both alleles while the proband IV-1 was homozygous T/T showing a novel homozygous abnormal splice variant c.4982+1G>T, p.Val1661fs in the MYFO gene.
The patient had homozygous abnormal splice variant in the Myoferlin gene while both the parents were heterozygous carriers ( Fig. 2). To our knowledge, this variant has not been previously reported in the literature. Furthermore, the variant is novel in gnomAD exomes and 1000 genomes. Furthermore, the results of in-silico tools, publicly and commercially available, used to aid in the interpretation of sequence variants identified in this study are presented in Table 1. ACMG criteria of variant classification PVS1 and PM2 suggest that the mutation in MYOF is pathogenic. Segregation of the variant also supports its deleterious nature.
The in-silico analysis performed in this study.
| S. no | Online tools | Pathogenicity score for variant in the MYOF gene c.4982+1G>T, p.Val1661fs |
|---|---|---|
| 1 | Mutation Taster | Disease causing |
| 2 | Polyphen-2 (v2.2.2, released in Feb, 2013) | 1.0 |
| 3 | Mutation Assessor 2.0 | 1.0 |
| 4 | Phylop (phyloP46way_placental) | 0.25 |
| 5 | VEST3 | 0.84 |
| 6 | CADD | 26.0 |
| 7 | Phastcons 1.4 | 1.0 |
| 8 | Exome aggregation consortium version 0.3.1 | 0.0% |
| 9 | 1000 genomes | 0.0% |
| 10 | SIFT | 0.090 |
| 11 | Diploid internal frequency | 0.0% |
DISCUSSION
We have reported a clinical case of an 18-month-old boy with sitting and standing difficulties suggesting FIS where WES identified a novel homozygous abnormal splice variant c.4982+1G>T, p.Val1661fs in the MYOF gene. This was further validated by Sanger sequencing, where the proband IV-1 showed homozygous mutation, and both parents were heterozygous at this position.
Ferlin family consists of several C2 domain proteins mediating vesicle fusion and membrane trafficking. MYOF and DYSF are expressed in different cells, including endothelial cells, lung’s epithelial cells and muscle cells; they both show a high structural homology ( Peulen et al., 2019). Despite these similarities, MYOF has a limited capacity to compensate for DYSF loss in dystrophic patients ( Inoue et al., 2006). Therefore, MYOF does not have an overlapping function with DYSF. MYOF is highly expressed in myoblasts before fusion to syncytial myotubes; however, in mature myotubes, their expression is decreased while DYSF is increased ( Davis et al., 2002). MYOF is also expressed following muscular injury and causes membrane repair and muscle regeneration through activating an NFAT-dependent promoter ( Demonbreun et al., 2010). Furthermore, loss of MYOF but not DYSF reduced the sensitivity of excitation–contraction coupling in mice skeletal muscle, producing small and disorganized ryanodine receptor cluster size relative to wild-type myofibers ( Barefield et al., 2021). These studies highlight the essential role of these proteins in muscle physiology, and emerging evidence has shown that disorders in the ferlin family proteins have a potential pathological role. Furthermore, DYSF gene mutation is linked to proximal and distal muscular weakness, Limb-Girdle muscular dystrophy 2B, and Miyoshi muscular dystrophy 1, respectively ( Liu et al., 1998). MYOF gene also has been shown to be linked with muscular dystrophy. Kiselev et al. (2019) reported a case of a woman with cardiomyopathy and limb-girdle muscular dystrophy with a truncated variant of myoferlin; two adjacent cis variants in MYOF heterozygosity was identified: a 1-bp deletion (c.2576delG, NM_133337) and a c.2575G-C transversion (c.2575G-C, NM_133337), which resulted in a frameshift mutation (Gly859GlnfsTer8) ( Kiselev et al., 2019). Similarly, in vitro studies showed that initial fusion events occur in MYOF null mice, but the later stages of myogenesis are defective; therefore, these mice display a dystrophic phenotype, and muscles fail to regenerate after injury ( Doherty et al., 2005). Recently, MYOF gene has been shown to be linked with other pathologies, such as angioedema and cancers ( Kiselev et al., 2019; Peulen et al., 2019; Santacroce et al., 2021).
In the current study, WES results, along with the presented clinical picture, were capable of categorizing the identified variant as pathogenic and stood along with the few studies that linked MYOF to human diseases. Our data identified for the first time a novel homozygous abnormal splice variant in MYOF gene which led to muscular hypotonia in an infant which might be FIS.
CONCLUSIONS
We are reporting a novel homozygous abnormal splice variant in the MYOF gene in a patient with generalized hypotonia, along with FIS. We suggest that screening of such families must be done by using WES to add new genes and novel mutations to the literature. Our study is a step forward and it will help to add to the literature of the diseases in Saudi population. Our study further supports the evidence of the MYOF gene being associated with a human disease phenotype and widens the field for advance research on ferlin protein family in connection with disorders related to the gene.