
INTRODUCTION
There is no health without mental health. In a recent report laid out by the World Health Organization on World Mental Health ( Kessler et al., 2006), the Kingdom of Saudi Arabia (KSA) holds the first position among the Gulf Cooperation Council states to promote a national research-based mental health survey, the Saudi National Mental Health Survey ( Almutairi, 2015). Mental disability in KSA is similar to that in the rest of the world, distressing personalities of all ages, cultures, and socio-economic statuses. For that reason, researchers are paying much emphasis on overcoming mental disabilities ( Alenezi et al., 2022). This will act as a new milestone to guide national mental health policy, treatment, and diagnosis in the Kingdom. The reason behind this mental disability arises from some genetic changes induced by environmental factors and oxidative stress ( Salim, 2014).
The mechanistic studies of small drug molecular systems, which can inhibit fibril formation in the brain’s central nervous system, are of prime importance at an early stage of intervention ( Sedov and Khaibrakhmanova, 2022). These effective tuning agents are capable of stabilizing the monomer by blocking the formation of toxic oligomers and distracting the monomeric protein to non-toxic intermediates (off-pathway) ( Kreiser et al., 2020). Small molecule candidates are being designed to inhibit the aggregation of Aβ, α-synuclein, and prions. Protein aggregation is the hallmark that is responsible for various neurodegenerative abnormalities inside the human body. Some of the associated disorders are Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease, and type II diabetes, ( Lee et al., 2011; Soto and Pritzkow, 2018; Monaco and Fraldi, 2020; Calabrese et al., 2022). However, the inner cellular environment does not support protein misfolding, as other proteins like chaperons prevent protein aggregation and assist in unfolding the misfolded polypeptide chains ( Mattoo and Goloubinoff, 2014; Koldewey et al., 2017). Despite all the efforts, some of the proteins remain in the aggregated form as their formation is induced by factors such as disease-related mutations and environmental conditions ( Chiti et al., 2002). This aggregation or misfolding of protein was also termed amyloid fibrils derived from the precursor proteins, which can be considered as a potential target for the control of amyloidosis ( Ow and Dunstan, 2014; Nyström and Hammarström, 2022). These amyloid fibrils are very well recognized by the presence of a cross β-sheet structure, 7-12 nm in size, mostly composed of hydrophobic amino acid residues ( Tjernberg et al., 2002; Rambaran and Serpell, 2008; Toyama and Weissman, 2011).
Numerous studies showed that amyloid fibrils accumulated inside the neurons causing neurological dysfunctions that lead to dementia or mental disability ( Benilova et al., 2012; DeTure and Dickson, 2019; Sehar et al., 2022). Therefore, inhibiting the formation of amyloid fibrils could be a promising approach for the design of therapeutic candidates. Many scientists have conducted research for the treatment of amyloid-related human diseases but problems associated with amyloid formation persist, and more advancement is required in this field ( Shewmaker et al., 2011).
In this regard, numerous neurological drugs were used in the treatment of neurodegenerative disorders. One of the well-known therapeutic drugs is rivastigmine ( Fig. 1), which is a noncompetitive carbamate acetylcholinesterase inhibitor that maintains the acetylcholine levels in the neurons ( Yang et al., 2013). Rivastigmine is administered to humans either orally or through a transdermal patch for the treatment of neurodegenerative diseases with moderate to severe symptoms. However, the effectiveness of this drug was reduced upon oral administration, as it is very difficult to cross the blood–brain barrier in the human body. Thus, it is better to adopt a carrier molecule for proper delivery or it must be given through the nasal route directly to the brain for proper bio-distribution and pharmacological properties ( Tenovuo, 2005; Gao et al., 2021). However, the problem related to the loss of neurons in the brain continues in patients, making it necessary to develop an approach to inhibit protein misfolding or amyloidosis. Therefore, it is important to recognize new targets for the proper functioning of neurodegenerative drugs to overcome amyloidosis ( Liu et al., 2016).

Human insulin (HI) is a model protein comprising two polypeptide chains (chain A and chain B) and plays a central role in the regulation of glucose metabolism ( Sędzikowska and Szablewski, 2021). Both chains are cross-linked via disulfide bonds ( Gangarde et al., 2021). The purpose of choosing insulin for this investigation is because it is more prone to aggregation in both monomeric and dimeric forms, which poses a challenge to its safe commercial use ( Gangarde et al., 2021). The literature revealed that HI is quite sensitive in vitro to various factors such as a change in pH and temperature and can form amyloid under these conditions. At higher temperatures and a physiological pH of 7.4, HI forms aggregates similar to amyloids ( Rahamtullah and Mishra, 2021). HI assists in misfolding and amyloid formation as confirmed by Congo red and Thioflavin T (ThT) dye-binding studies ( Duboisset et al., 2013; Yakupova et al., 2019). However, HI itself is a very stable protein and is resistant to aggregation in vivo due to its α-helical structure and disulfide bridges. Nevertheless, under in vitro conditions, it is easily susceptible to forming amyloid-like structures ( Zheng and Lazo, 2018). As the accumulation of amyloids will result in neuron loss, inhibition of amyloid formation directly protects against the progression of neurodegenerative disorders such as AD, PD, and type II diabetes ( Stroo et al., 2017). In this context, many studies were performed, which showed the inhibitory potential toward amyloid aggregation using various drugs or natural products such as quercetin ( Wang et al., 2011), gallic acid ( Jayamani and Shanmugam, 2014), rosmarinic acid (( Zheng and Lazo, 2018), peptides, and cucurbituril ( Bailey et al., 2012; Gangarde et al., 2021). However, the study on the inhibitory potential of rivastigmine against HI aggregation remains, which might also result in the removal of amyloid plaques in the brain.
Herein, we demonstrate the inhibition of amyloid formation using the rivastigmine drug. Nowadays, protein misfolding and aggregation are a wide field of research. Due to a lack of knowledge on the mechanism of protein–ligand interaction, misfolding, folding, inhibition, and aggregation of protein aggregates, there is no permanent cure for such types of diseases ( Chaudhuri and Paul, 2006; Rahman et al., 2022). Therefore, in the present study, we perform a comprehensive analysis on the inhibition of insulin fibrillation by rivastigmine using various spectroscopic and biophysical techniques. Furthermore, we also investigate the interaction between the protein and ligand using computational methods.
MATERIALS AND METHODS
Materials
Rivastigmine, HI, ThT, and 1-anilino-8-naphthalenesulfonate (ANS) were procured from Sigma-Aldrich, USA. 4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid (HEPES) buffered saline (pH 7.4), procured from Sigma, USA, was used. Analytical standard solvents were procured from Fisher Scientific, UK. Milli-Q ® water was used throughout the studies.
Sample preparation
HI was dialyzed in 50 mM HEPES buffer, which was subjected to size exclusion chromatography prior to use. Insulin concentration was determined spectrophotometrically considering that the molar extinction coefficient (ɛ) at 280 nm is 5,734 M −1 cm −1 (using a Perkin–Elmer lambda 25 double beam UV–vis spectrophotometer). Protein samples containing NaCl (10 μM) were placed at elevated temperature (65 °C) in parallel sets to obtain the fibrils of insulin. Any effect exerted by rivastigmine was analyzed using several biophysical techniques after 72 h of incubation at 65 °C.
pH measurements
A benchtop pH meter (Mettler Toledo; S400) pH/mV and temperature meter were used to adjust the pH of the biological solution in an elevated temperature range. The reading was repeated three times with the standard buffer, and the average of these was taken as the final value.
Turbidity
The turbidity assay is frequently exploited to study protein aggregation behavior. Turbidity leads to a decrease in the intensity of the light that passes through a turbid solution due to light scattering. Turbidity measurements of HI (μM) incubated without and with rivastigmine in an increasing manner were carried out using an absorption spectrometer (Hitachi 2900) at a fixed wavelength of 350 nm in a cuvette of 1 cm path length ( Zhao et al., 2016; Huang et al., 2021).
ThT fluorescence
ThT is a benzothiazole dye that binds with the aggregates of HI protein with the cross-β structure of amyloid fibrils. Upon binding, it gives enhanced fluorescence at 480 nm when excited at 440 nm. A solution of ThT solution (50 μM) was prepared in HEPES buffer (50 mM, pH 7.4) and its concentration was determined using a Shimadzu UV–visible spectrophotometer by using an extinction coefficient ɛ M=26,600 M 1cm −1 at 412 nm. For this purpose, we added ThT to the aliquots of HI and HI containing different amounts of rivastigmine and subjected them to incubation for 15 min under dark conditions ( Krebs et al., 2005; Kroes-Nijboer et al., 2009; Rane et al., 2021). The assay solution was excited at 440 nm and the emissions were measured over the range of 400 to 700 nm using a fluorescence spectrophotometer (Shimadzu RF6000) equipped with a Julabo Eyela water circulator. The slit widths were kept at 5 nm for both excitation and emission.
Rayleigh light scattering
The light scattering method is used to analyze the size and gross conformation of a protein in solution because the intensity of light scattered by the solution in terms of the time-averaged intensity ( Zakariya et al., 2020; Rane et al., 2021) in the absence and presence of varying concentrations of rivastigmine with respect to the native protein was examined using the Raleigh light scattering study. In this case, aliquots of the reaction mixture were excited at 350 nm, and then emission intensity was recorded at 300 to 400 nm using a Shimadzu (RF–5301 PC) fluorescence spectrophotometer with a path length of 1 cm. The excitation and emission slits were fixed at 5 nm. Each sample was run in triplicate and the data were averaged for plotting scattering intensity with respect to time.
ANS fluorescence assay
A polarity-sensitive fluorescent dye, ANS, was employed to measure the exposure of hydrophobic groups during protein aggregation. A stock solution of ANS was incorporated into each aliquot (2.5 mL) of the HI sample (in both the absence and presence of rivastigmine) so that the final ANS concentration in each aliquot was 30 μM ( Nielsen et al., 2001; Rane et al., 2021). The concentration of ANS was determined using the molar extinction coefficient (ε 350 = 5000 M −1 cm −1) ( Rane et al., 2021). After adding ANS to the samples in a molar ratio, the samples were kept in the dark for 30 min at room temperature. The fluorescence spectra were recorded using a Shimadzu RF-5301 PC with excitation fixed at 380 nm and emissions were monitored in the range of 390-650 nm. Slit widths were set at 5 nm for both excitation and emission. Each spectrum was blank-corrected. Data points were the average of triplicate measurements.
Far-UV circular dichroism (CD) measurements
To assess the secondary structural changes that rivastigmine might induce on HI, far-UV CD spectra (190 to 250 nm) were analyzed at 25 °C using a Jasco Spectropolarimeter (J-815) (Jasco, Tokyo, Japan) using a regular rectangular quartz cuvette with a 1 cm path length and at a scan speed of 100 nm min −1. Appropriate baseline corrections were implemented against the buffer solution to extract the corrected CD spectra. A curve-fitting program CDNN 2.1 software determines the percent of secondary structures present in protein under different conditions.
Molecular docking
AutoDock vina software was employed to perform molecular docking studies to gain insights into the binding sites of Aβ-42 and acetylcholinesterase (AChE) with rivastigmine ( Trott and Olson, 2010). The structures of Aβ-42 [protein data bank (PDB) ID: 2MXU] and AChE (PDB ID: 4EY7) were obtained from the research collaboratory for structural bioinformatics (RCSB) protein data bank (PDB). Four units of Aβ-42 and one monomeric subunit of the AChE structure were considered for docking. Protein preparation including the addition of polar hydrogens and assign Kollman charges was performed in MGLTools–1.5.6. The grid-center was adjusted to x = −16.747, y = 6.113, and z = 7.492 with a grid-box dimension of 46×44×52 Å 3 and x = −24.688, Y = 22.433, and Z = 0.664 having a grid-box dimension of 68×76×66 Å 3 to cover the residues of Aβ-42 and AChE, respectively with a spacing of 1 Å. The structure of rivastigmine (CID: 77991) was obtained from the PubChem database in the sdf format. The structure of rivastigmine was minimized and saved in the pdb format using Chimera-1.10.2 ( Pettersen et al., 2004) prior to docking. Docking was performed using the Broyden–Fletcher–Goldfarb–Shanno algorithm in Autodock vina. The exhaustiveness (number of independent runs) was set to 100 and the remaining parameters were set to their default values. An root mean square deviation (RMSD) tolerance of 2 Å was considered while performing the cluster analysis. The 2D interaction of the docked complex was prepared using the Accelrys Discovery Studio Visualizer 2.1 (Accelrys, San Diego, CA, USA). The 3D visualization of the residues of the docked pose was analyzed using Chimera-1.10.2 ( Pettersen et al., 2004).
Molecular dynamics simulation
Molecular dynamics (MD) simulation of the docked-out complexes with Aβ-42 and AChE showing the best binding affinity (more negative) was performed in an aqueous environment. MD simulations were performed using the Gromacs–2018.1 packages using the AMBER99SB–ILDN force field ( Van Der Spoel et al., 2005). The topology of the rivastigmine molecule was generated in Antechamber using the AM1–BCC charge model by using AmberTools21 ( Sousa da Silva and Vranken, 2012). Moving forward, the Aβ-42, AChE, and the docked complex were solvated using TIP3P water in a triclinic box and the respective systems were neutralized by the addition of an equal number of counter Na +/Cl − ions followed by energy minimization to a maximum of 5000 steps using steepest descent minimization in order to remove the steric clashes. The first equilibration of all systems [NVT equilibration: In the canonical ensemble, amount of substance (N), volume (V) and temperature (T) are conserved] was done using a V-rescale thermostat at 300 K and constant volume for 200 ps at a coupling constant of 0.1 ps ( Bussi et al., 2007). The second equilibration [NPT equilibration: In the canonical ensemble, amount of substance (N), volume (V) and temperature (T) are conserved] was performed using Parrinello–Rahman barostat at 1.0 bar and 300 K for 200 ps having a coupling constant of 2 ps ( Parrinello and Rahman, 1981). A cutoff distance of 1.4 nm was set for the Coulombs and Lennard Johns interaction having an integration time step of 2 fs ( Darden et al., 1993). Particle Mesh Ewald governed the electrostatic interaction and Fourier transformation had a grid spacing of 0.16 nm ( Essmann et al., 1995). Production MD ran for 150 ns for Aβ-42 and 100 ns for AChE alone and for the docked complex it was performed by generating 15,000 frames with Aβ-42 and 10,000 frames with AChE for each MD trajectory. The MD trajectories were subjected to PBC corrections and Gromacs utilities were used to perform the analysis. The molecular mechanics poisson-boltzmann surface area (MM–PBSA) calculations were performed for calculating the binding energies involved in the interaction of docked complexes over the course of MD simulation.
RESULTS AND DISCUSSION
Turbidity measurement
The turbidity (“haze development”—the scattering of light from fine protein aggregates) assay is a convenient way to study the aggregated proteins ( Steiner et al., 2010; Oukarroum et al., 2012). It is evident that most proteins exhibited a peak at about 280 nm due to the presence of a chromophoric amino acid group (tyrosine and tryptophan), which cuts off above 320 nm ( Rane et al., 2021). However, aggregation of proteins leads to the broadening of the peak rather than cutting off because of the turbidity ( Zhao et al., 2016). If the turbidity is detected by the eye (in the visible spectrum), the sample is highly aggregated ( Kosinski-Collins, 2003). This is because of the fact that the intensity of scattered light is higher at low wavelengths as it is inversely proportional to the fourth power of wavelength (λ nm) ( Kitchener et al., 2017). An increase in turbidity is associated with an increase in light scattering in the UV range, which is an indication of aggregate formation ( Oukarroum et al., 2012). Thermal aggregation in insulin protein does not give rise to a visibly turbid solution as reported for other proteins and thus shows lower absorbance values. However, when compared to HI protein at 25 °C, the insulin protein preincubated at 65 °C (using water bath) for 72 h displays a substantial increase in turbidity. In general, the aggregated solution gives more absorbance value due to the unfolding of the polypeptide chain. Successive addition of rivastigmine to a fixed amount of protein causes a steady decrease in turbidity and thereby absorbance, even at a higher temperature (65 °C) ( Fig. 2), which is recognized as a reduction in the number or size of protein aggregates. This is mainly because rivastigmine stabilizes the protein system through intermolecular interaction between them.

Effect of rivastigmine on insulin fibrillation
ThT, a benzothiazole dye, is the most extensively used fluorophore to examine the fibrillation behavior of proteins ( Biancalana and Koide, 2010). In general, ThT shows enhanced fluorescence upon binding to the cross-β sheet structure, thus allowing the quantification of amyloid fibrils ( Biancalana and Koide, 2010). In our case, we have studied the inhibitory potential of rivastigmine with insulin fibrillation ( Fig. 3). It is evident that HI exhibits fluorescence at room temperature; however, it has been found to increase upon incubation at 65 °C for 72 h, indicating the presence of aggregates ( Rane et al., 2021). This increase in emission profile is related to the fact that intermolecular interactions among polypeptides are weakened at higher temperatures, which causes unfolded protein conformation suitable for the nucleation step for aggregation ( de Groot et al., 2007). Therefore, we subjected the heat-treated (65 °C) insulin protein to different concentrations of rivastigmine after incubation for 72 hr. Spectrum analysis revealed that there was a decrease in fluorescence intensity with respect to the increase in the concentration of the drug, indicating the inhibitory potential of rivastigmine ( dos Santos Rodrigues et al., 2023). The possible inhibition or suppression of fibril formation is related to the fact that hydrogen bonding and other weak interaction are involved between rivastigmine and the amino acid of the polypeptide which lead to photoelectron transfer in the excited state ( Iannuzzi et al., 2015). The probable interaction has been further corroborated with molecular docking and MD studies. Hence, we speculate that the insertion of the rivastigmine molecule into HI may stabilize the polypeptide and thereby make it more resistant to extreme conditions.

Rayleigh light scattering measurements
To obtain a clearer picture of the inhibitory potential of rivastigmine on insulin fibrillation, the inhibition of fibrillation behavior was also examined in the absence and presence of varying concentrations of rivastigmine with the help of Rayleigh light scattering (RLS) measurement ( Alsenaidy, 2018). The data were collected after incubation of insulin protein solution at 65 °C for 72 h to detect the onset of aggregation. In general, insulin protein showed fluorescence maxima at 350 nm upon excitation of a protein sample at the same wavelength in buffer solution. Hence, the analysis of the emission profile at 350 nm becomes a promising tool to dictate protein aggregation leading to a rise in the RLS data. Figure 4 displays a change in scattering intensity measured after the heat-treated insulin sample at pH 7.4. The results displayed an increase in scattering intensity than native insulin, suggesting the aggregate formation after thermal treatment. This means that the structure of a protein is lost and it forms maximum aggregates under the influence of thermal denaturation. With the successive addition of rivastigmine under amyloidogenic conditions, there is a decrease in the fluorescence intensity implying dose-dependent inhibition of protein aggregate formation. The lowering in RLS intensities could be speculated that the presence of rivastigmine may stabilize the polypeptide through pi (π)–π intercalation and hydrophobic interactions.
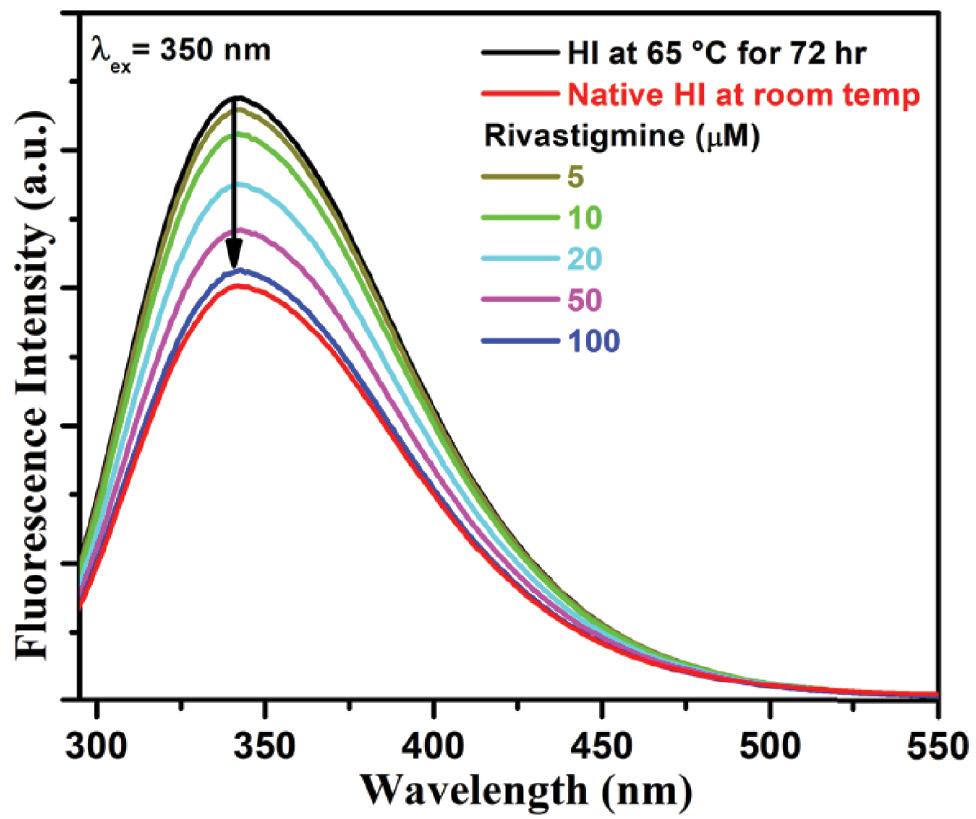
RLS fluorescence intensity spectra of HI at room temperature and at 65 °C for 72 h incubated along with different concentrations of rivastigmine (5-150 μM) in 50 mM HEPES buffer. The samples were excited and emitted at 350 nm. Data are presented as the mean of three different experiments performed in triplicate. Abbreviations: HI, human insulin; RLS, Rayleigh light scattering.
Monitoring the degree of surface hydrophobicity
ANS is a polarity-sensitive fluorescent probe that binds to the exposed hydrophobic region of the protein, ultimately resulting in an increase of fluorescence intensity ( Semisotnov et al., 1991; Guliyeva and Gasymov, 2020). During amyloid fibrillation, the partial unfolding and misfolding of protein lead to the exposure of hydrophobic areas, and this exposition acts as a template for further fibril polymerization ( Kundu et al., 2020). This means that when fibril elongation takes place, more binding sites for ANS will be available ( Cardamone and Puri, 1992). Hence, it is widely used to analyze the proteins’ folding/unfolding processes, which are mainly governed by electrostatic and hydrophobic interactions. Figure 5 depicts the enhancement in the ANS fluorescence spectrum of thermally exposed insulin at around 473 nm upon excitation at 380 nm. This is because of the disruption of weaker interactions. However, the addition of a different amount of rivastigmine to the ANS fluorescence primarily treated with thermally incubated protein caused successive decreases in emission intensity with a significant red shift of 10 nm.
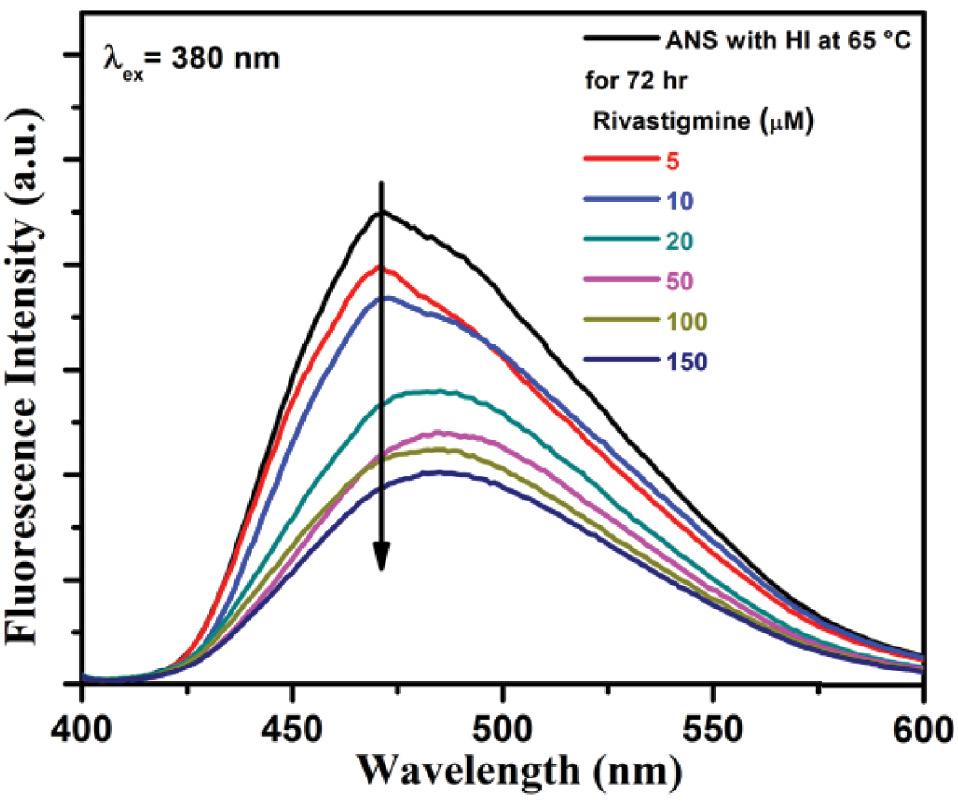
ANS fluorescence spectra of insulin incubated at 65 °C for 72 h in the absence and presence of rivastigmine (5-150 μM). Excitation wavelength was 380 nm and emission ranged from 400 to 600 nm. Each spectrum represents an average of three scans. Abbreviation: ANS, 1-anilino-8-naphthalenesulfonate.
The results showed that the addition of rivastigmine to the system represented an inhibitory agent, which may be augmented by the fact that π–π intercalation between the aromatic rings of rivastigmine may stabilize the polypeptide. Therefore, the intercalation of rivastigmine may have resulted in less exposure to buried hydrophobic residues, thereby stabilizing the protein, which is very important to protect the brain-related disease (AD and PD). The molecular action of rivastigmine is associated with the prevention of misfolding of proteins, and hence several compounds in this line have been tested to treat neurodegenerative disease.
Effect of rivastigmine on the secondary structure of insulin during thermal incubations
To investigate the effect of rivastigmine on the secondary structural transformation in the aggregation process of HI, the Far-UV CD technique has been carried out in the region 190-250 nm ( Fig. 6). Native insulin protein presented two negative bands at 208 nm and 215 nm, which are the characteristic features of a typical α helical protein. At room temperature (25 °C), rivastigmine hardly induces any change in the ellipticity and shape of the spectra and hence the secondary structure of the protein ( Fig. 6). However, thermal incubation of native protein at 65 °C for 72 h leads to the generation of a new peak at around 218 nm, and minima at 208 and 222 nm were reduced indicating β-sheet-rich structure formation of the polypeptide. Hence, the transition of α conformation to β-sheeted structures has been observed at higher temperatures. It is interesting that the conformation of α-helical was retained when insulin protein was incubated under similar conditions in the presence of rivastigmine. The presence of rivastigmine has prevented the appearance of β-sheeted conformations because the ellipticity obtained was not at around 218 nm but it resembled 208 nm and 222 nm conformation. Thus, the results suggest that rivastigmine has the potential to stabilize a protein in its native state thus preventing it from fibrillation.

In silico studies
Molecular docking with amyloid β (Aβ-42) and AChE inhibitors
In silico molecular docking analyses were carried out to validate our experimental results The Gibbs free energy value computed for the Aβ-42–rivastigmine complex was determined to be −6.2 kcal/mol. Out of the 10 binding modes acquired from docking, the 3D depiction of the best conformational mode having the best binding energy is shown in Figure 7a. Multimeric chains (four units) of the Aβ-42 protein were considered for the docking and simulation studies. The binding constant of the Aβ-42–rivastigmine complex was calculated to be 3.5 × 10 3 M −1. The rivastigmine molecule forms two hydrogen bonds with His14 (chain C) and Gly33 (chain A) residues of the Aβ-42 protein as shown in Figure 7b. The residues Val12, His14, Ile32, and Leu34 of chain A, Val12, Leu17, Ile32, Gly33, and Leu34 of chain B and Leu17, Ile32, and Gly33 of chain C are involved in hydrophobic and van der Waal interactions as depicted in Figure 7b. The aromatic ring of rivastigmine forms π–cation interaction with the His14 residue of chain B of Aβ-42. Overall, the binding of rivastigmine with Aβ-42 is significantly favored and stabilized by hydrogen bonding, hydrophobic, van der Waals, and π–cation interactions. Moreover, there are no D chain residues of Aβ-42 interacting with rivastigmine.
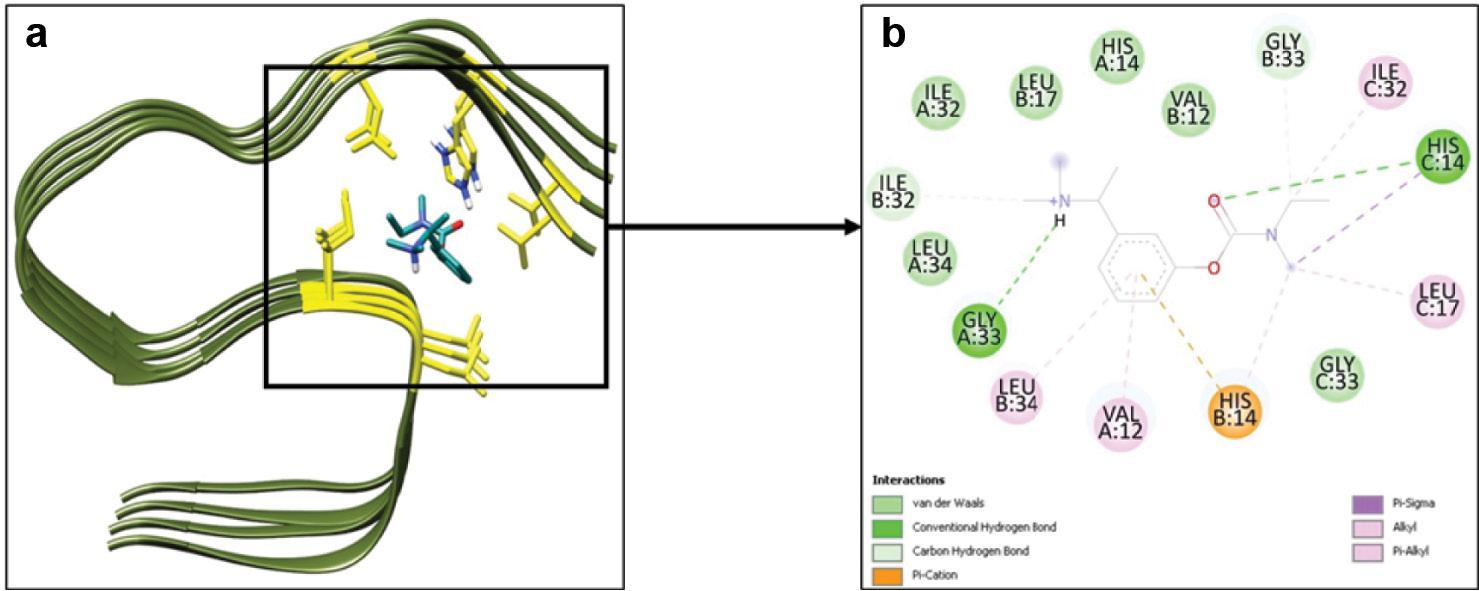
(a) 3D representation of the biomolecular complex of Aβ-42 (PDB ID: 2MXU) complexed with rivastigmine. (b) 2D depiction of the residues of Aβ-42 involved in interaction with rivastigmine.
Similarly, the Gibbs free energy value computed for the AChE–rivastigmine complex was determined to be −7.1 kcal/mol. Out of the 10 binding modes acquired from docking, the 3D depiction of the best conformational mode having the best binding energy is shown in Figure 8a. The binding constant of the AChE–rivastigmine complex was calculated to be 1.6 × 10 5 M −1. Rivastigmine forms one hydrogen bond with the Tyr337 residue of AChE as shown in Figure 8b. The aromatic ring of rivastigmine is involved in Π–Π stacking with the Trp86 (also forms hydrophobic interactions) and Tyr337 residues of AChE. The residues Tyr72, Asp74, Gly82, Thr83, Asn87, Gly121, Tyr124, Ser125, Ser203, Tyr341, Trp439, His447, Gly448 and Tyr449 possess van der Waals nature of interaction with rivastigmine. In addition, Glu202 of AChE is involved in π–cation interaction with the rivastigmine’s positively charged nitrogen atoms. Therefore, the interaction or binding of rivastigmine with AChE is significant and favored and stabilized by various nonbonded interactions including hydrogen bonding, and hydrophobic, van der Waals, and π interactions.

MD simulations
Root mean square deviation (RMSD) and root mean square fluctuation (RMSF)
The analysis of the MD simulation trajectories of Aβ-42 and Aβ-42–rivastigmine complex was performed using various Gromacs utilities. Initially, the RMSD of the backbone atoms of Aβ-42 alone and in complex with rivastigmine was determined with respect to their respective initial conformations as shown in Figure 9. The RMSD of the backbone atoms of Aβ-42 alone and Aβ-42–rivastigmine complex attained equilibrium after around 100 ns of and were stable over the remaining course of MD simulation. The average RMSD values of Aβ-42 and Aβ-42–rivastigmine complex were calculated to be 0.32 and 0.38 nm, respectively. As evident from Figure 9, the binding of rivastigmine does not induce any significant changes in the conformational structure of Aβ-42. The RMSD values of Aβ-42 and Aβ-42–rivastigmine were found to be in an admissible range of below 0.5 nm. The RMSD values of Aβ-42 in the presence of rivastigmine confer equilibrated and durable stability of the Aβ-42–rivastigmine complex.
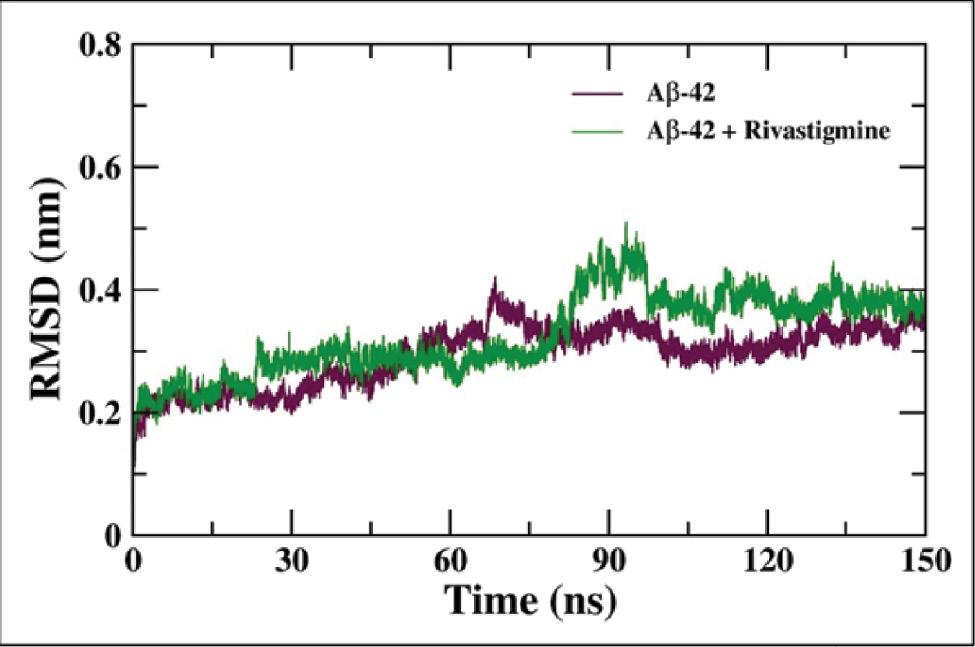
RMSD of the backbone atoms of Aβ-42 alone and the Aβ-42–rivastigmine complex over the course of 10 ns of MD simulation. Abbreviation: MD, molecular dynamics.
Furthermore, the slight fluctuations in the RMSD of the backbone atoms of Aβ-42 can be further explained through the residue-wise RMSF analysis of Aβ-42 and Aβ-42–rivastigmine complex. The residue-wise RMSF of the backbone atoms of Aβ-42 illustrates comparatively increased fluctuations in the residues (20-30) which are characteristic of the loop region of the protein. In addition, it is also evident from Figure 10 that the binding of rivastigmine with Aβ-42 incites the decrement of RMS fluctuations of the Aβ-42 residues. Overall, the residual fluctuations of the backbone atoms of Aβ-42 are conciliated upon binding with rivastigmine.
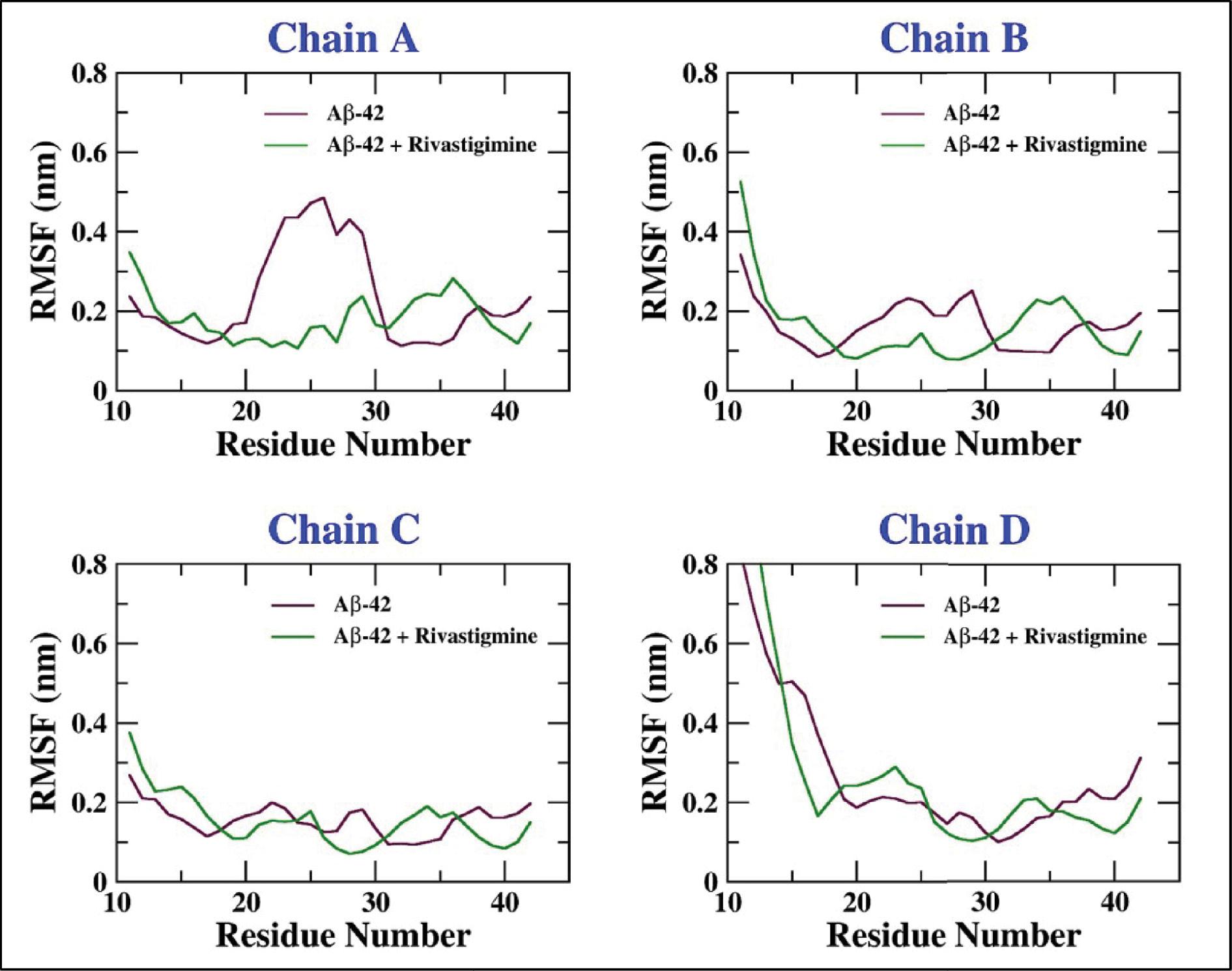
Residue-wise RMSF of the backbone atoms of Aβ-42 alone and the Aβ-42–rivastigmine complex.
The analysis of the MD simulation trajectories of AChE and AChE–rivastigmine complex was performed using various Gromacs utilities. Initially, the RMSD of the backbone of AChE alone and the AChE–rivastigmine complex was determined with respect to their respective initial conformations as shown in Figure 11a. The RMSD of the backbone atoms of AChE alone and the AChE–rivastigmine complex after 25 ns of equilibration was stable over the remaining course of MD simulation. The average RMSD of AChE and the AChE–rivastigmine complex were calculated to be 0.17 and 0.16 nm, respectively. As evident from Figure 11a, the binding of rivastigmine induces insignificant changes in the conformational structure of AChE. RMSD values below 0.3 nm are acceptable and appreciable ( Nour et al., 2022; Hashmi et al., 2023). The RMSD values of AChE in the presence of rivastigmine confer equilibrated and durable stability of the AChE–rivastigmine complex.
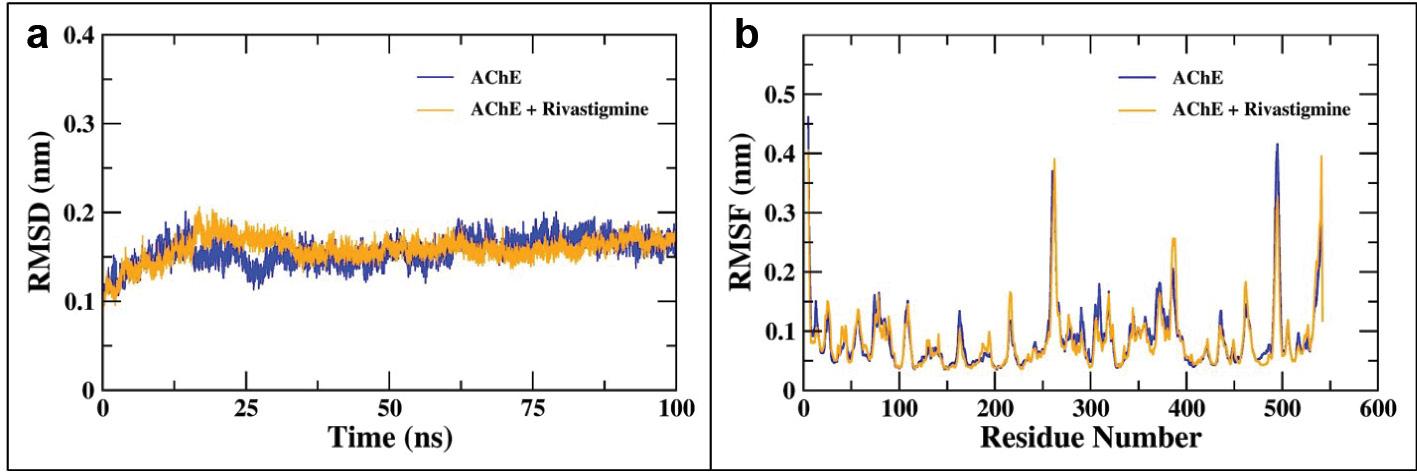
(a) RMSD of the backbone atoms of AChE alone and the AChE–rivastigmine complex over the course of 100 ns of MD simulation. (b) Residue-wise RMSF of the backbone atoms of AChE alone and the AChE–rivastigmine complex. Abbreviations: AChE, acetylcholinesterase; MD, molecular dynamics.
Furthermore, the RMSF analysis was performed, the results of which are presented in Figure 11b. The residue-wise RMSF values of the backbone atoms of AChE and the AChE–rivastigmine complex for the majority of residues were below 0.3 nm. However, the increased fluctuations in the residues (257-260 and 496-498) are accounted for due to the loop region of the protein. The binding of rivastigmine leads to slight decrement in the fluctuations in the 496-498 residues of the AChE protein. Briefly, the residue-wise RMSF plot of the backbone atoms of AChE describes no noticeable alterations in the residual fluctuations upon binding with rivastigmine.
Analyzing the Rg, solvent accessible surface area, and energies of the system
Furthermore, the analysis of the MD simulation trajectories was proceeded by assessment of an important parameter known as the radius of gyration (R g) which is the RMS distance (mass–weighted) of atoms from their common center of mass ( Ahmad et al., 2021). The radius of gyration (R g) analysis endorses the structural stability of Aβ-42 and Aβ-42–rivastigmine complex by evaluating the structural compactness. The Rg plot of the backbone atoms of Aβ-42 and Aβ-42–rivastigmine complex shown in Figure 12a suggests stable structural dynamics. The average Rg values for Aβ-42 alone and the Aβ-42–rivastigmine complex were found to be 1.38 and 1.41 nm, respectively. The insignificant change in the Rg values upon binding of rivastigmine with Aβ-42 depicts structural integrity conferring stability in the dynamics of complex structures over the course of MD simulation with the evolution of time ( Khamouli et al., 2022).
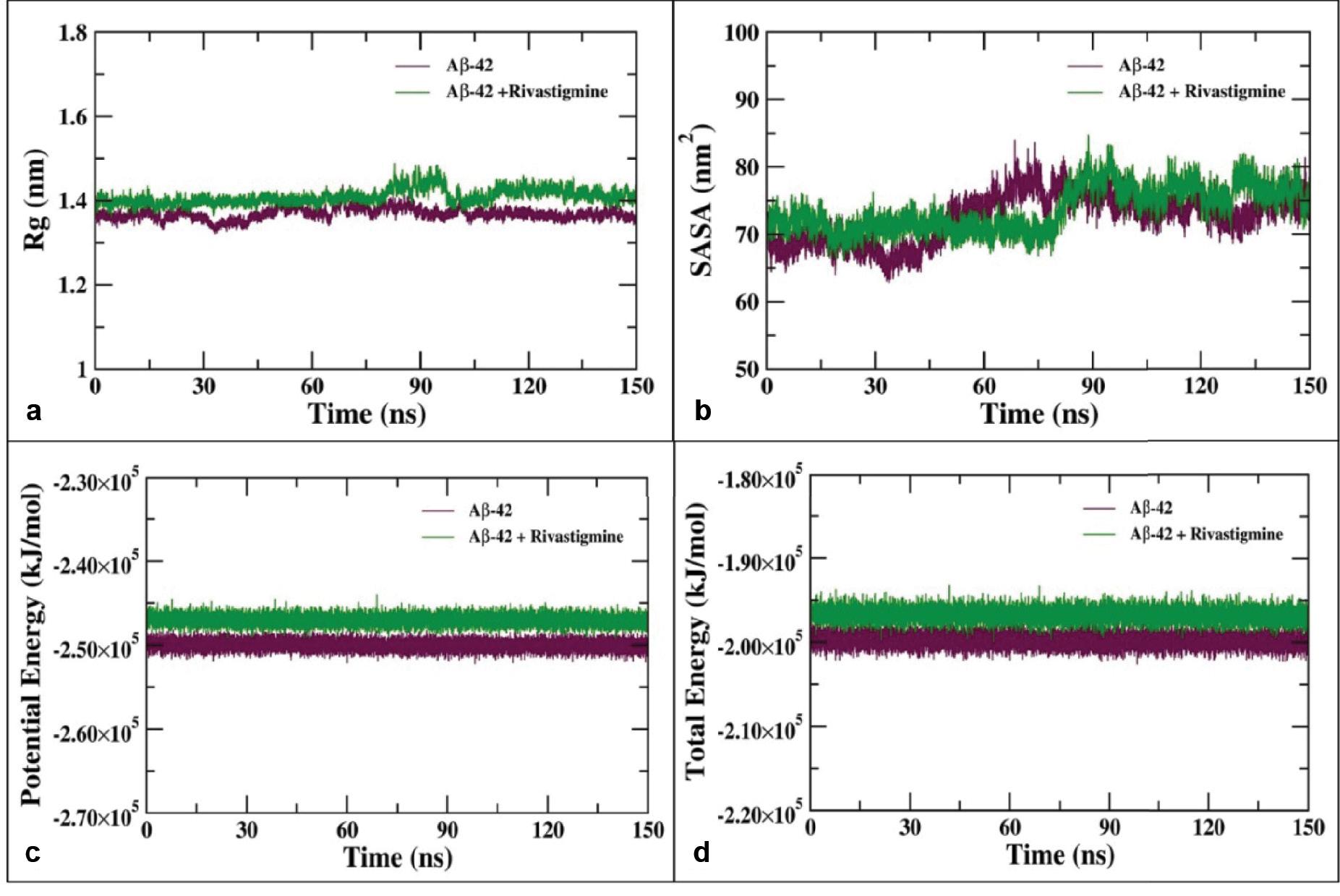
(a) Radius of gyration (Rg) of the backbone atoms of Aβ-42 alone and in complex with rivastigmine during 150 ns of MD simulation. (b) SASA of Aβ-42 and the Aβ-42–rivastigmine complex as a function of the evolution of time. (c) Potential and (d) total energies of Aβ-42 and Aβ-42–rivastigmine complex systems over the course of 100 ns of MD simulation. Abbreviations: MD, molecular dynamics; SASA, solvent accessible surface area.
The solvent accessible surface area (SASA) was analyzed using the gmx sasa utility to determine the effect of rivastigmine binding with Aβ-42 and accessibility of the protein–ligand complexes with the solvent molecules ( Ouassaf et al., 2022). The SASA was calculated for the 100 ns simulation trajectories of Aβ-42 and its complex with rivastigmine as shown in Figure 12b. The average SASA values for Aβ-42 alone and Aβ-42–rivastigmine complex were estimated to be 75.82 and 77.27 nm 2, respectively.
The binding of rivastigmine with Aβ-42 does not result in major changes in the SASA values of Aβ-42 suggesting that the Aβ-42–rivastigmine complex possesses stable nature of interaction and there is no significant alteration in the conformations of Aβ-42 over the entire course of MD simulation upon binding with rivastigmine. The potential and total energies of Aβ-42 and Aβ-42–rivastigmine complex systems were evaluated and found to be stable over the entire course of MD simulation as a function of time as shown in Figure 12c and d.
The stability of proteins undergoing MD simulation is assessed by one of the important parameters known as the radius of gyration (R g) which is the RMS distance (mass–weighted) of atoms from their common center of mass ( Ahmad et al., 2021). The radius of gyration (R g) analysis endorses the structural stability of AChE and AChE–rivastigmine complex by evaluating the structural compactness. The Rg of the backbone atoms of AChE and AChE–rivastigmine complex shown in Figure 13a indicates the stable structural dynamics of AChE alone and in complexes with rivastigmine. The average Rg values for AChE alone and AChE–rivastigmine complex were found to be 2.31 and 2.28 nm, respectively. A slight decrement in the Rg values upon binding of rivastigmine with AChE illustrates structural integrity and stable structural dynamics over the course of 100 ns of MD simulation ( Khamouli et al., 2022).
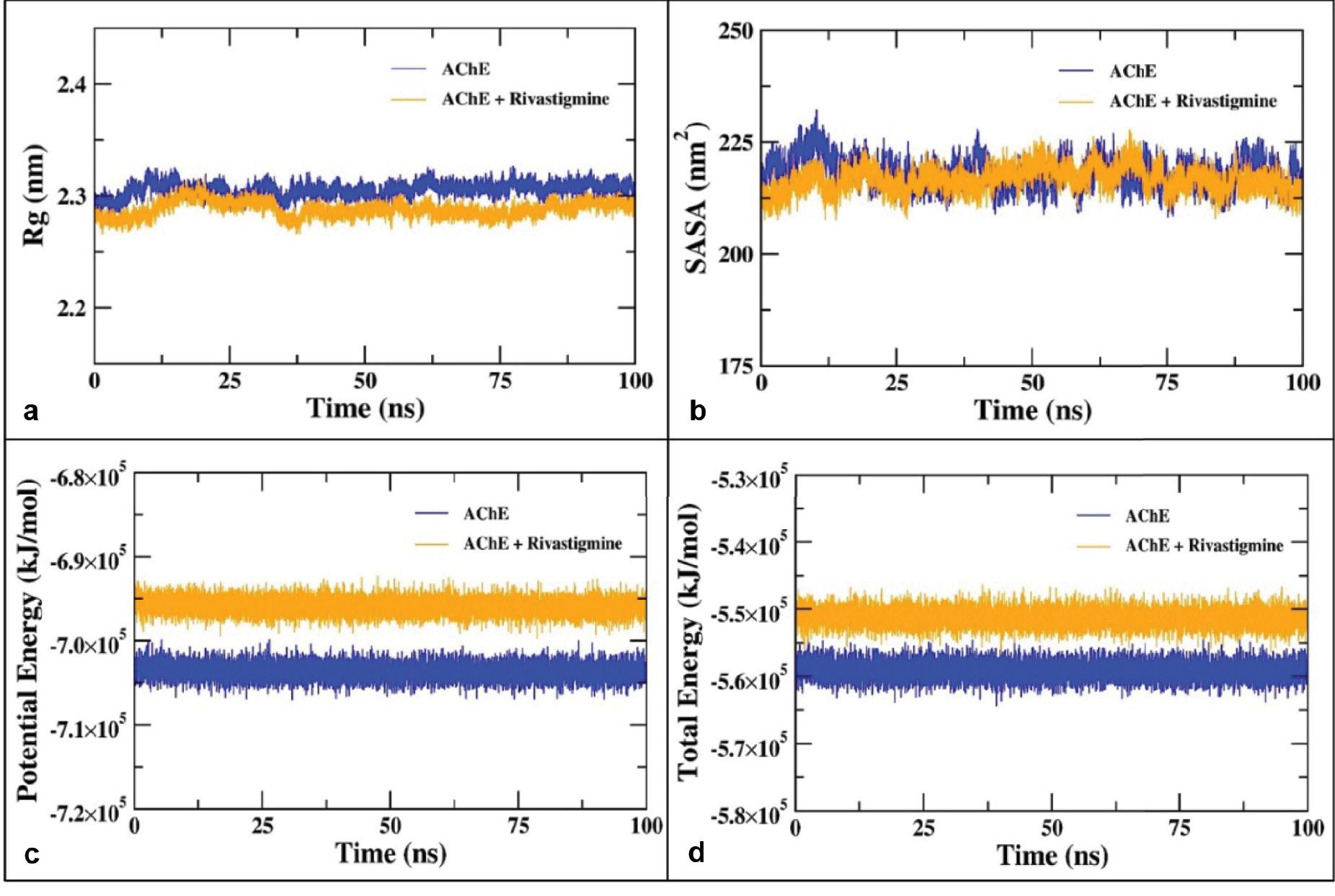
(a) Radius of gyration (Rg) of the backbone atoms of AChE alone and in complex with rivastigmine during 100 ns of MD simulation. (b) SASA of AChE and the AChE–rivastigmine complex as a function of the evolution of time. (c) Potential and (d) total energies of AChE and AChE–rivastigmine complex systems over the course of 100 ns of MD simulation. Abbreviations: AChE, acetylcholinesterase; MD, molecular dynamics; SASA, solvent accessible surface area.
SASA was analyzed using the gmx sasa utility to determine the effect of rivastigmine binding into the hydrophobic core and accessibility of AChE to the solvent molecules ( Ouassaf et al., 2022). The SASA was calculated for the 100 ns simulation trajectories of AChE alone and AChE–rivastigmine complex as shown in Figure 13b. The average SASA values for AChE alone and the AChE–rivastigmine complex were estimated to be 222.38 and 219.15 nm 2, respectively. The binding of rivastigmine with AChE does not result in major changes in the SASA values of AChE suggesting that rivastigmine binding possesses stable nature of interaction without remarkable alterations in the conformations of AChE over the entire course of MD simulation. The potential and total energies of AChE and AChE–rivastigmine complex systems were evaluated and found to be stable over the entire course of MD simulation as a function of time as shown in Figure 13c and d ( Khamouli et al., 2022).
Hydrogen bond analysis
The interaction of rivastigmine with Aβ-42 protein was analyzed by determining the percent contribution of the residues of Aβ-42 forming H-bond with rivastigmine. The hydrogen bond existence map of the Aβ-42–rivastigmine complex over the course of 150 ns of MD simulation is shown in Figure 14. The higher propensity for hydrogen bonds formed between the protein–ligand complexes is relatable to the number of polar groups in the ligand molecule ( Quds et al., 2022; Hashmi et al., 2023). As evident from the data, there are four major hydrogen bond interactions over the course of the MD simulation. The existence of hydrogen bonds between rivastigmine and Aβ-42 was found to be significant due to the interaction of Glu11, Val12, and His13 residues of Aβ-42 protein. The Glu11 residue of Aβ-42 is involved in bidentate hydrogen bond formation with the rivastigmine molecule. As shown in Figure 14, both the sidechain oxygen atoms (OE1 and OE2) of Glu11 are involved in hydrogen bond formation with the nitrogen atom (N1) of rivastigmine having a percent existence of 14.76% between GLU11(OE1):: LIG(N1) and 21.07% between GLU11(OE2):: LIG(N1). Similarly, the percentage contribution of the Val12 and His 13 residues is 7.63 and 6.24%, respectively. The interaction of solvent molecules at the binding site results in the weakening of the hydrogen bond formation, due to which few other residues in the binding vicinity had an existence of less than one percent ( Ding et al., 2015).
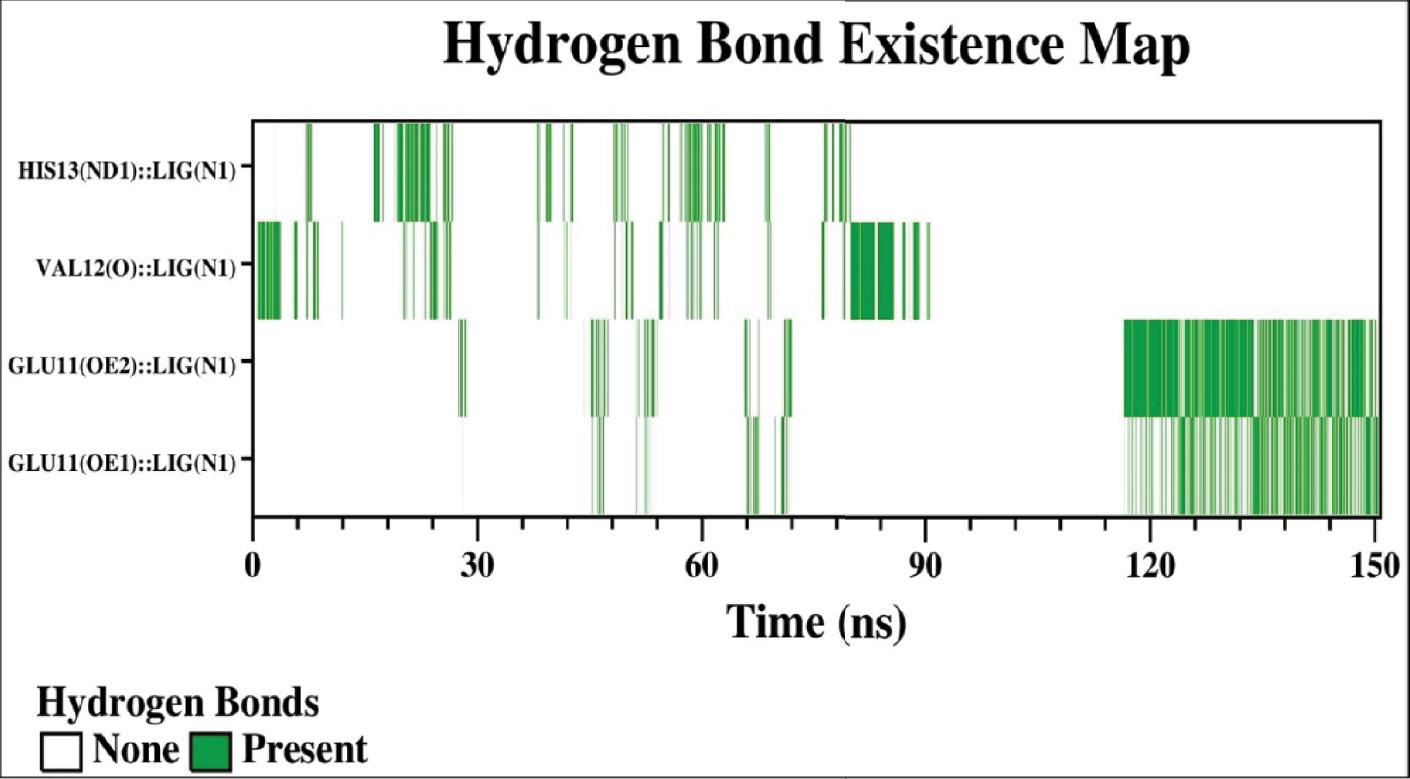
Representation of the hydrogen bond that exists between the atoms of the residues of Aβ-42 and the atoms of rivastigmine as a function of time.
The interaction of rivastigmine with AChE was analyzed by determining the percent contribution of the residues of AChE forming H-bond with rivastigmine. The hydrogen bond existence map of the AChE–rivastigmine complex over the course of 100 ns of MD simulation is shown in Figure 15. The more the number of polar groups present in the ligand molecule interacting with the protein, the higher the propensity for the persistent hydrogen bonds formed between the protein–ligand complexes ( Quds et al., 2022). As evident from the data, there are three major hydrogen bond interactions over the course of the MD simulation. The existence of hydrogen bonds between rivastigmine and AChE was found to be significant due to the interaction of Glu202 and Tyr337 residues of AChE. The percent contribution of the AChE residues involved in interaction with rivastigmine as shown in Figure 15 was analyzed and it was observed that both the sidechain oxygen atoms (OE1 and OE2) of Glu202 are involved in hydrogen bonding with the nitrogen atom (N1) of rivastigmine having a percent existence of 90.33% between GLU202(OE1):: LIG(N1) and 24.65% between GLU202(OE2)::LIG(N1), thus causing Glu202 to form a bidentate hydrogen bond with rivastigmine molecule. Similarly, the hydrogen atom of the OH group of the sidechain of Tyr337 is involved in hydrogen bond formation with the oxygen atom (O2) of rivastigmine showing 91.52% between TYR337(OH):: LIG(O2). A few residues in the binding vicinity had H-bond existence of <5% existence. The interaction of solvent molecules at the binding site results in the weakening of the hydrogen bond formation ( Ding et al., 2015).

Analysis of energy contributions through MM–PBSA calculations
The contribution of binding energies involved in the interaction of Aβ-42 with rivastigmine was determined using MM–PBSA calculations. The MM–PBSA calculations were performed for 500 frames uniformly extracted from the last 100-150 ns of MD simulation trajectories of the Aβ-42–rivastigmine complex. Noncovalent forces are prominent in the interactions of proteins and ligands. These forces are characterized as electrostatic forces, van der Waals forces, hydrophobic interactions, and hydrogen bonds imparting positively or negatively into the overall binding of the ligands with the proteins ( Hashmi et al., 2023). The magnitude of binding energies arising due to the interaction of the Aβ-42–rivastigmine complex is listed in Table 1. The binding of rivastigmine to Aβ-42 is significantly favored by electrostatic and van der Waals forces. However, the SASA energy also had slight contributions to the binding; meanwhile, the polar solvation energy hinders the binding of rivastigmine with Aβ-42. The contrary effect of polar solvation energy is due to the interference of the continuum solvent into the interaction of the Aβ-42–rivastigmine complex. The relative binding energy of the Aβ-42–rivastigmine complex was calculated to be −185.747 ± 1.286 kcal mol −1.
Binding free energy (kcal mol −1) for the interaction of rivastigmine with Aβ-42 using MMBSA analysis.
| Parameters | Aβ-42–rivastigmine complex |
|---|---|
| ΔE vdW | −111.245 ± 0.654 |
| ΔE ele | −268.778 ± 1.393 |
| ΔE PSE | 208.439 ± 2.254 |
| ΔE SASA | −14.220 ± 0.075 |
| ΔE BE | −185.747 ± 1.286 |
Abbreviations: ΔE BE, binding energy; ΔE ele, electrostatic energy; MMBSA, molecular mechanics with generalised born and surface area solvation; ΔE PSE, polar solvation energy; ΔE SASA, solvent accessible surface area energy; ΔE vdW, van der Waals energy.
The contribution of binding energies involved in the interaction of AChE with rivastigmine was determined using MM–PBSA calculations. The MM–PBSA calculations were performed for 500 frames uniformly extracted from the last 50 ns of MD simulation trajectories of the AChE–rivastigmine complex. Noncovalent forces are prominent in the interactions of proteins and ligands. These forces are characterized as electrostatic forces, van der Waals forces, hydrophobic interactions, and hydrogen bonds imparting positively or negatively into the overall binding of the ligands with the proteins ( Hashmi et al., 2023). The magnitude of binding energies arising due to the interaction of the AChE–rivastigmine complex is listed in Table 2. The binding of rivastigmine to AChE is found to be most favored by electrostatic and moderately by van der Waals forces. The SASA energy also had minute contributions to the binding; however, the polar solvation energy remarkably hinders the binding of rivastigmine to AChE. The contrary effect of polar solvation energy is due to the interference of the continuum solvent with the interaction of the AChE–rivastigmine complex. The relative binding energy for AChE–rivastigmine interaction was calculated to be −346.674 978 ± 0.696 kcal mol −1.
Binding free energy (kcal mol −1) for the interaction of rivastigmine with AChE using MMBSA analysis.
| Parameters | Aβ-42–rivastigmine complex |
|---|---|
| ΔE vdW | −145.004 ± 0.522 |
| ΔE ele | −523.891 ± 0.723 |
| ΔE PSE | 339.371 ± 0.636 |
| ΔE SASA | −17.124 ± 0.032 |
| ΔE BE | −346.674 ± 0.696 |
Abbreviations: AChE, acetylcholinesterase; ΔE BE, binding energy; ΔE ele, electrostatic energy; ΔE PSE, polar solvation energy; ΔE SASA, solvent accessible surface area energy; ΔE vdW, van der Waals energy.
CONCLUSION
In summary, we reported the inhibitory potential of a neurological-based rivastigmine drug candidate against model proteins, i.e. HI, employing a biophysical tool. For this purpose, several examinations like ThT assay, measurement of hydrophobicity, Rayleigh scattering measurements, and turbidity measurement pertaining to neurodegenerative disease were performed. The ThT assay exposed that protein causes decrease in turbidity and thereby absorbance in the presence of increasing amounts of rivastigmine, which is documented as a reduction in the number or size of protein aggregates. This is mainly because protein gets stabilized with rivastigmine through intermolecular interaction between them. Decrease in the fluorescence intensity measured in RLS and ANS measurements suggested the inhibitory action of rivastigmine toward protein aggregation. The far-UV CD results revealed that rivastigmine stabilized a protein in its native state. Furthermore, molecular docking and MD simulation of rivastigmine reveal its proficient binding with amyloid β (Aβ-42) and AChE inhibitors with the vital active site residual. The present study provides a mechanistic insight into the inhibition of amyloid fibrillation by rivastigmine, which offers evidence to expand various formulations in the pharmaceutical industry to prevent amyloid fibrillation.