
INTRODUCTION
Intellectual disability (ID; also known as mental retardation) is a major public health issue that affects up to 2% of the population and is considered to have an intelligence quotient of <70 and deficits in adaptive behaviors that affect normal life. ID is subdivided into nonsyndromic and syndromic forms depending on the clinical features of the patients ( Ropers, 2008; Mefford et al., 2012).
There are almost 700 identified rare genetic diseases linked with different forms of IDs and can be mitochondrial, autosomal dominant, autosomal recessive, or X-linked (Orphanet data base). However, autosomal recessive forms of IDs are rare and account for <12% of cases of total IDs ( Jamra, 2018). Moreover, autosomal recessive is most common in consanguineous communities as in the Middle East ( Mir et al., 2009; Mochida et al., 2009; Philippe et al., 2009; Abou Jamra et al., 2011; Kakar et al., 2012; Marangi et al., 2013; Giorgio et al., 2016; Abbasi et al., 2017; Liang et al., 2020).
Autosomal recessive intellectual developmental disorder 13 (OMIM: 613192) is a rare disorder caused by a homozygous mutation in the trafficking protein particle complex subunit 9 ( TRAPPC9) gene ( Mir et al., 2009; Mochida et al., 2009). The disease is caused by the loss-of-function mutations in any of 23 exons of the TRAPPC9 gene located on chromosome 8q24.3 ( Mir et al., 2009). Previous articles reported >55 cases of the disease with most of them being from Middle Eastern consanguineous families ( Amin et al., 2022). The disease is clinically characterized mainly by ID and developmental delay with microcephaly and brain abnormalities with some patients having dysmorphic facial features and obesity ( Yousefipour et al., 2021). A novel homozygous c.1422C>T transition in the TRAPPC9 gene, resulting in a protein change p.R475X mutation, was reported in affected members with mental retardation from a large consanguineous family from Pakistani ( Mir et al., 2009). Similarly, a homozygous mutation p.R475X was identified in affected members of a large consanguineous Syrian family with autosomal recessive mental retardation. The patients had severe motor delay, neonatal hypotonia, single words or no speech, walking between ages 5 and 7 years, stereotypic movements, growth retardation, and hand flapping. The affected member also showed dysmorphic features, synophrys, including low frontal hairline, along with microcephaly 9 ( Abou Jamra et al., 2011). Furthermore, a consanguineous family from Tunisia with three brothers having autosomal recessive intellectual developmental disorder-13 (MRT13; 613192), with a homozygous mutation c.1708C>T, p.R570X exon 9 of the TRAPPC9 gene was reported by Philippe et al. (2009).
Homozygous mutations in the PLOD3 gene were reported to cause an autosomal recessive connective tissue disorder (OMIM: 612394). The first report of the disease was published by Salo et al. (2008), who reported that a female born to a nonconsanguineous couple of European descent showed a compound heterozygous mutation c.668A>G resulting in the amino acid change to p.Asn223Ser, and one nucleotide deletion c.2071 delT resulting in a translational frameshift and generating a premature stop codon (p.Cys691AlafsX9) in the sequence. Subsequent families with consanguineous members have a homozygous PLOD3 c.809C>T; p.(Pro270Leu) variant. The PLOD3 gene was further characterized the clinical features, which included joint contractures, low bone mineral density and fractures, scoliosis, myopia, sensorineural deafness, dysmorphic features, skin and nail abnormalities, and rupture of a vascular aneurysm ( Ewans et al., 2019; Vahidnezhad et al., 2019). A recent study showed a homozygous mutation of c.1216_1218delCTC (p.L406del) in PLOD3, which was inherited from heterozygous parents ( Zhou et al., 2022).
This study reports the clinical and molecular characteristics of a young female who had whole exome sequencing (WES) and found to have two novel homozygous variants in the PLOD3 and TRAPPC9 genes leading to bone fragility with contractures and intellectual developmental disorder.
METHODS
Ethical approval and sample collections
We took ethical approval for this study from the committee of the Center of excellence in Genomic Medicine Research, King Abdulaziz University Jeddah, (013-CEGMR-02-ETH) for this study. The experimental study was done according to the international guidelines of the Declaration of Helsinki 2013. Blood samples from which DNA was extracted was taken after having consent from the family and the DNA was stored in EDTA tubes (Roche Life Science) as previously reported ( Naseer et al., 2021). Nanodrop™ 2000/2000c spectrophotometers were used for the measurement of the concentration of DNA (Thermo Fisher Scientific Waltham, MA, USA).
Clinical details of the patient
In this study, a consanguineous family with an affected girl having ID, developmental delay, microcephaly, spastic contractures, exaggerated deep tendon reflexes, and positive clonus bilaterally syndrome was recruited. The family with one affected girl and two normal bothers along with the parents were studied ( Fig. 1). The parents were first degree cousin and had three kids, two normal male while one affected girl with ID, developmental delay, microcephaly, spastic contractures, exaggerated deep tendon reflexes, and positive clonus bilaterally syndrome along with dysmorphic facial features. All members of the family were examined clinically and further WES was performed for the affected proband.

Whole exome sequencing
WES technique was used to find the mutation leading to the disease in the affected member of a Saudi family. One affected member’s DNA sample was used for WES by latest Illumina NextSeq 550 by using a High-Output v2 kit, and 2 × 76 paired-end reads were sequenced on an Illumina NextSeq as previously explained ( Naseer et al., 2020; 2021). Quality control was maintained on Illumina sequencing platforms along with DNA templates were constructed for quantitation.
For WES, SureSelect XT_V6_PostCap libraries (Agilent Technologies, USA) were constructed using 2 μg of genomic DNA of the family members. The target regions with an average throughput depth of 130 bp and 100 bp paired-end reads were sequenced on a HiSeq 2500 system (Illumina, USA). These variants were filtered based on different parameters such as genomic position, protein effect, quality, frequency, pathogenicity, and former associations with the phenotype. Various bioinformatic analyses were carried out to identify causative variants linked to the disease. Raw data FASTQ files were aligned using the BWA Aligner ( http://bio-bwa.sourceforge.net/). Copy number variation and insertion/deletion (InDel) detection were performed using SAMTOOLS ( http://samtools.sourceforge.net/). The sequence reads were mapped against the hg19 (NCBI build GRCh37) human reference sequence ( http://genome.ucsc.edu/) and compared against sequences in the dbSNP ( http://www.ncbi.nlm.nih.gov/snp/) and the 1000 Genomes Project databases ( http://www.1000genomes.org/data).
WES sequencing were obtained in the FASTQ files format to convert to BAM files from Illumina machines and finally the BAM files were converted to variant call format files for analysis. Bioinformatics Illumina tools called “bcl2fastq v2.20.0” ( https://www.bioinformatics.babraham.ac.uk/projects/fastqc) were used. Furthermore, various free online available analysis tools were used for the identification of the causative variants linked with the disease such as pathogenicity, homozygosity, heterozygosity, rare/novel (MAF+0.01%) frequency, genomic position, functional status using PolyPhen/SIFT predicted damage, damaging effect of the protein that shows linkage with the disease and its phenotype. Single nucleotide polymorphisms (SNPs) or variants at this phase were identified at nucleotide resolution. SNPs recognized were contrasted with genomAD databases ( https://gnomad.broadinstitute.org/), SnpEff ( http://snpeff.sourceforge.net/SnpEff.html), and 1000 genome ( https://www.internationalgenome.org/). Bioinformatics tools were used by applying various filters. The GRCh37 database was used for alignment of the reference sequence. Allele frequencies filtered <5.0% whereas the Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/), and nonsense, frameshift, and splice-site variants in disease-related genes with a minor allele frequency ≤1.0% were observed in gnomAD. American College of Medical Genetics (ACMG) and American College of Pathologists along with multiple lines of computational evidence supporting the deleterious effect on the gene or gene product, such as conservation, evolutionary, and splicing impact, were studied as previously done by Abdulkareem et al. (2023). Variants were reported according to the HGVS nomenclature ( https://varnomen.hgvs.org/). Furthermore, various in silico tools were used to identify the missense variants to predict amino acid change in the protein structure, such as SIFT ( http://swissmodel.expasy.org), PolyPhen-2 software ( http://genetics.bwh.harvard.edu/pph2). Standard guidelines of the ACMG were followed. Mutation Tester ( http://www.mutationtaster.org/) was used for the prediction of pathogenicity, phyloP ( https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4702902/), 1000 Genomes database ( http://www.internationalgenome.org/), and phyloP. SIFT ( http://sift.bii.a-star.edu.sg/), CADD ( https://cadd.gs.washington.edu/), SiPhy ( https://omictools.com/siphy-tool), GERP++ ( http://mendel.stanford.edu/SidowLab/downloads/gerp/), PhastCons ( http://compgen.cshl.edu/phast/), and Exome Aggregation Consortium ( http://exac.broadinstitute.org/) were used.
Sanger sequencing
The results of WES showed novel variants that were further verified by using the Sanger sequencing technique for all other family members. For PCR and sequencing the targeted primer was designed by using an online primer 3 program, and the designed primer sequences were used as a forward primer PLOD3-F-5-GTAACGGGGCTGGGGATAG-3, reverse, PLOD3-R-5′-GTTGACCTCGAGACTCCCAG-3′ while for other forward primer for TRAPPC9-F-5-GAATGAATCAGCGAGTGGGT-3 and reverse primer for TRAPPC9-R-5-CTGACCCCAAAGCCCACC-3. Sequencing data files were obtained from the AB1 sequencing unit, and the obtained files were aligned with the reference sequence using the BioEdit software as shown in Figures 2 and 3.

Representative electropherogram of the TRAPPC9 gene. Sanger sequencing results showing that the parents III-1 and III-2 and the two normal boys are heterozygous carriers having G/A on both alleles while the proband IV-2 were homozygous A/A at the same position of c.3211G>A, p.G1071S in exon 22 of the TRAPPC9 gene. Abbreviation: TRAPPC9, trafficking protein particle complex subunit.
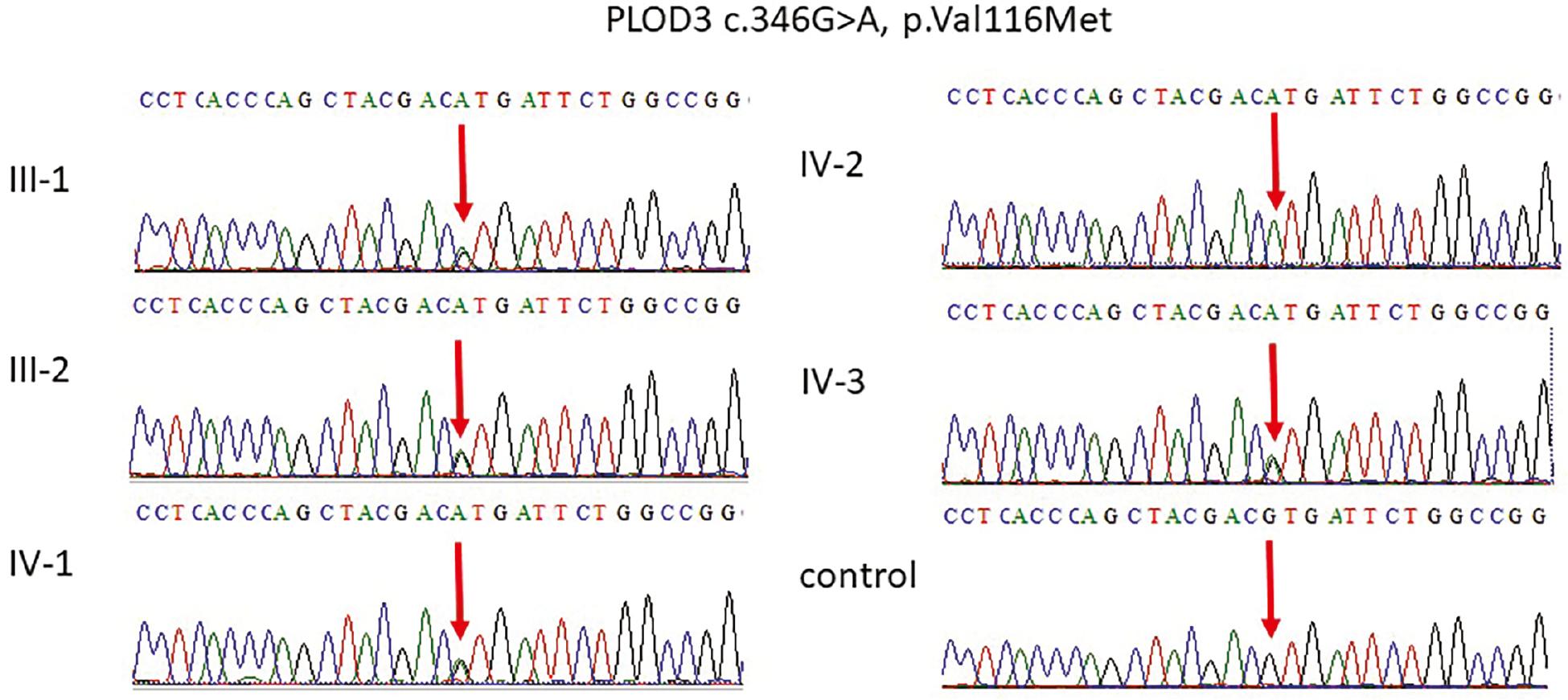
Representative electropherogram of the PLOD3 gene. Sanger sequencing showing that the parents III-1 and III-2 and two normal IV-1 and IV-3 members are carriers having G/A on both alleles while the proband IV-2 were homozygous A/A at the same position c.346G>A, p.V116M in exon 4 of the PLOD3 gene representing the diseased phenotype.
RESULTS
Case report
The girl was a product of a twin pregnancy from a consanguineous family born at 30 weeks of gestation with a birth weight of 1.5 kg and admitted to the neonatal intensive care unit for prematurity, respiratory distress (required mechanical ventilation), and jaundice (required phototherapy). Cranial ultrasound at the age of 8 months was within normal limits. Examination at the age of 2 years showed developmental delay, microcephaly, spastic contractures all over, exaggerated deep tendon reflexes, and positive clonus bilaterally, as shown in Figure 4. A repeat examination at the age of 9 years showed the same findings with the addition of understanding simple commands, being able to communicate basic needs, and having a happy disposition with normal body weight. MRI of the brain showed marked thinning of the corpus callosum and T2-weighted white matter hyperintensities in the centrum semiovale as well as periventricular and peritrigonal regions bilaterally. Nerve conduction studies and electromyography were within normal limits.
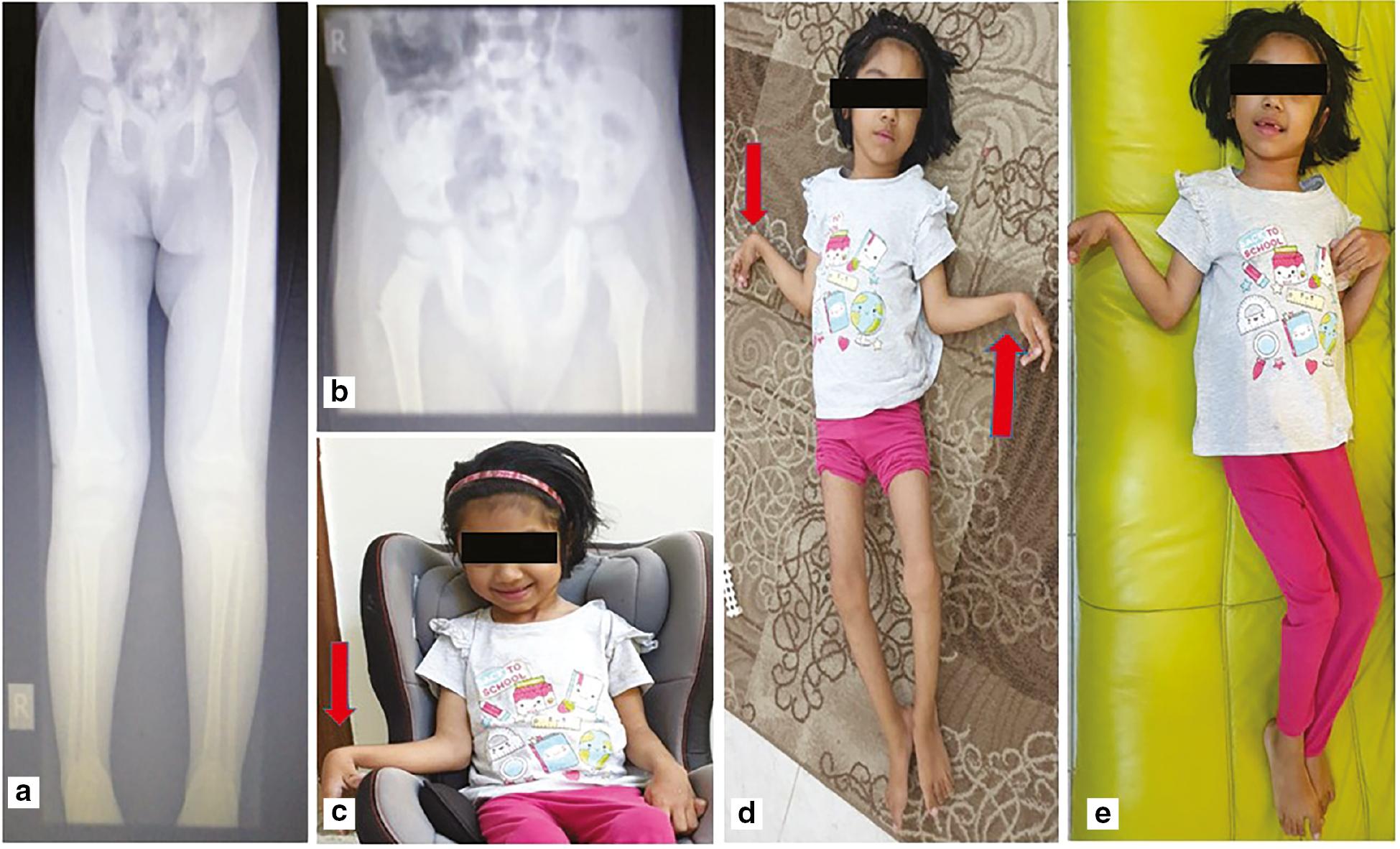
Clinical and radiological features of the affected individual. (a, b) Patient X-ray images showing extensive upper and lower extremity contractures with bones with no fractures. (c-e) The right and left hands with flexion contractures of proximal interphalangeal joint, flexion contractures of both elbows and all interphalangeal joints, worst on left hand fingers.
Whole exome sequencing
Our WES results showed the missense variant c.3211G>A, p.G1071S in the exon 22 of TRAPPC9 (NM_001160372.3) chr8:NC_000008.10:g.140744290C>T has not been reported previously as a pathogenic variant nor as a benign variant, to our knowledge. Both the parents are heterozygous carriers while the affected member is homozygous for this position. The two normal brothers are also carriers of this mutation. The missense p.G1071S variant is predicted to be damaging by both SIFT and PolyPhen2. The glycine residue at codon 1071 of TRAPPC9 is conserved in all mammalian species. The nucleotide c.3211 in TRAPPC9 is predicted conserved by GERP++ and phyloP across 100 vertebrates. For these reasons, the identified variant was classified as an uncertain significance.
The other missense variant c.346G>A, p.V116M in PLOD3 (NM_001084.4) chr7:NC_000007.13:g.100859600C>T was also identified that has not been reported previously as a pathogenic variant nor as a benign variant, to our knowledge. We also identified that both the parents are heterozygous carriers while the affected member is homozygous for this position. The two normal brothers are also carriers of this mutation. The p.V116M variant is observed in 3/30,616 (0.0098%) alleles in individuals from South Asian background in gnomAD Exomes and is novel (not in any individuals) in 1000 Genomes. The missense p.V116M variant is predicted to be damaging by both SIFT and PolyPhen2. The valine residue at codon 116 of PLOD3 is conserved in all mammalian species. The nucleotide c.346 in PLOD3 is predicted to be conserved by GERP++ and phyloP across 100 vertebrates as shown in Table 1.
Showing the details of the in silico analysis done in this study.
| S. No | Online tools | Pathogenicity score for variant in the TRAPPC9 gene c.3211G>A, p.G1071S | Pathogenicity score for variant in PLOD3 c.346G>A, p.V116M |
|---|---|---|---|
| 1 | MutationTaster | Disease causing | Disease causing |
| 2 | Polyphen-2 (v2.2.2, released in February 2013) | 1.0 | 1.0 |
| 3 | Mutation Assessor 2.0 | 1.03 | 1.0 |
| 4 | phyloP (phyloP46way_placental) | 0.72 | 0.23 |
| 5 | VEST3 | 0.82 | 0.90 |
| 6 | CADD | 21.0 | 25.0 |
| 7 | Phastcons 1.4 | 1.0 | 1.0 |
| 8 | SiPhy 0.5 | 15.0 | 12.0 |
| 9 | Exome Aggregation Consortium Version 0.3.1 | 0.0% | 0.0% |
| 10 | 1000 Genomes | 0.0% | 0.0% |
| 11 | Diploid internal frequency | 0.0% | 0.0% |
| 12 | SIFT | 0.09 | 0.07 |
Abbreviation: TRAPPC9, trafficking protein particle complex subunit.
Sanger sequencing
After the results of WES the identified mutation was segregated in all the family members as shown in figure to further validate the results. Sanger sequencing results showing that parents, III-1 and III-2, and two normal boys, IV-1 and IV-3, are carriers having G/A on both alleles while the proband IV-2 were homozygous A/A at the same position of c.3211G>A, p.G1071S in the exon 22 of the TRAPPC9 gene as shown in Figure 2. Furthermore, the Sanger sequencing was also done for the validation of the PLOD3 gene. The parents, III-1 and III-2, and two normal, IV-1 and IV-3, members are carriers having G/A on both alleles while the proband IV-2 were homozygous A/A at the same position c.346G>A, p.V116M in the exon 4 of the PLOD3 gene, representing the diseased phenotype as shown in Figure 3.
DISCUSSION
The human brain is a complex organ and its development is very complex that is the result of a cascade of reactions that are organized by a substantial number of genes ( Bae et al., 2015). In the current study, we are reporting two novel homozygous mutations in TRAPPC9 and PLOD3 genes causing ID microcephaly, included joint contractures, low bone mineral density, and dysmorphic feature in the affected member of the family. The most reliable clinical presentations of patients with TRAPPC9-related autosomal recessive ID were delayed speech development and cognitive impairment. Many patients were reported with other metabolic abnormalities and behavioral disorders, such as obesity and autism. However, dysmorphic features were also reported in some of the cases.
TRAPPC9 is a gene known for ID disorder and involved in the neuronal NF-kappa-B signaling pathway in the brain ( Hu et al., 2019). Previously, p.R475X a truncating mutation in the exon 7 of the TRAPPC9 gene was reported in a Pakistani family with ID ( Mir et al., 2009). Many other variants of this gene linked with ID have also been reported in different populations as well, for example, a French patient showed a compound heterozygous mutation inherited from the father along with that maternally inherited in exons 18 and 19 in TRAPPC9 ( Mortreux et al., 2018; Hnoonual et al., 2019; Wilton et al., 2020). So far TRAPPC9 reported mutations are loss-of-function mutations (nonsense, frameshift, splice site, or inframe insertions/deletions), and no genotype–phenotype correlation was identified ( Fig. 5).
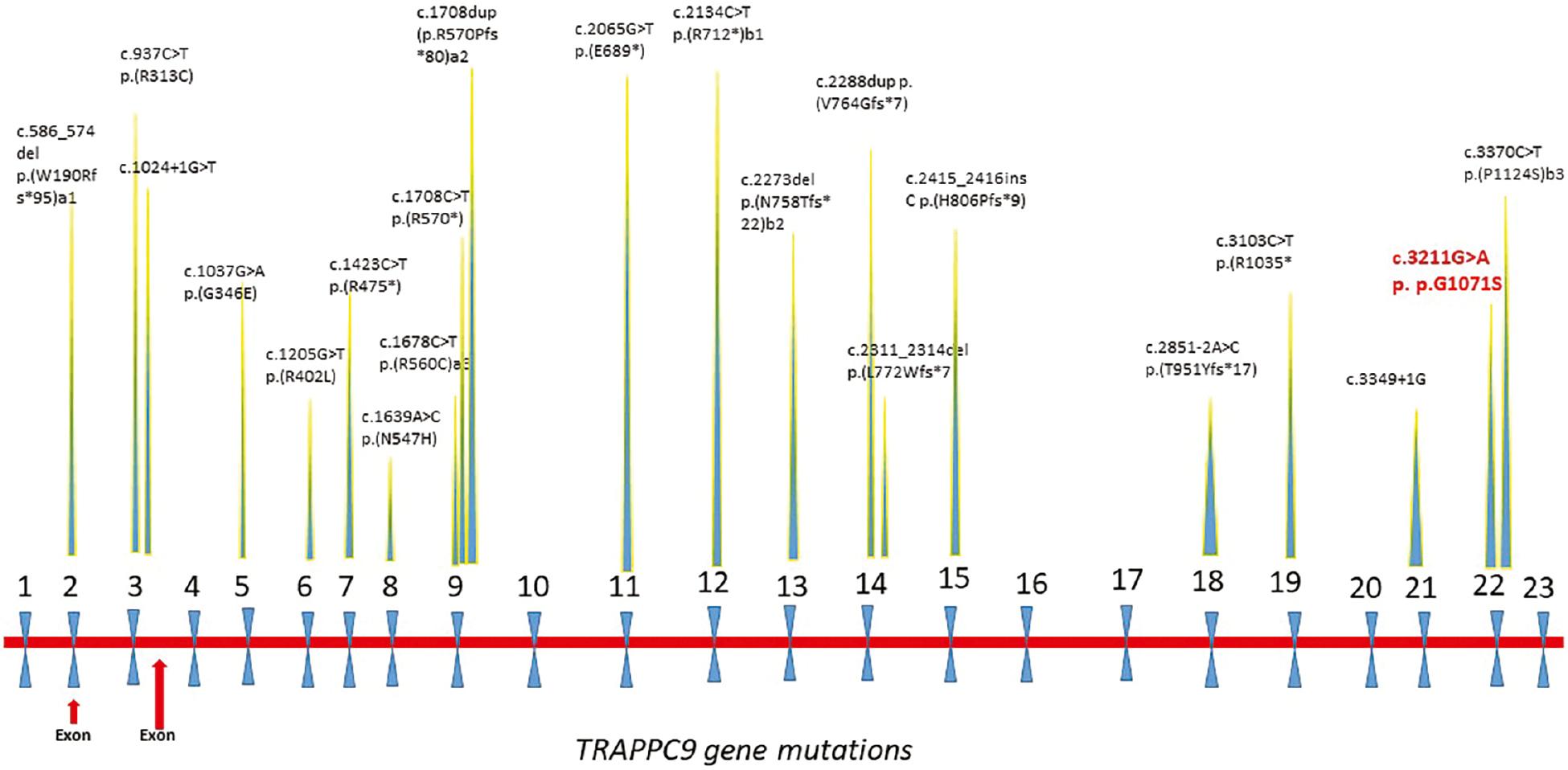
Reported mutations in the TRAPPC9 gene (NM_031466.7 transcript) known so far. Our reported mutation is shown in red. (a1, b1, a2 and b2) Representing the compound heterozygous mutations. Abbreviation: TRAPPC9, trafficking protein particle complex subunit.
TRAPPC9 is a 23-exon gene located in chromosome 8q24.3 that interacts with IKK-b and the NFkB-inducing kinase in addition to its role in NFkB signaling. It encodes a protein that plays important roles in brain development and regulation of neurogenesis. Loss-of-function mutations in this gene believed to impair NFkB signaling in neurons leading to ID ( Kakar et al., 2012). TRAPPC9-induced microcephaly is thought to arise from defects in neuronal differentiation ( Mochida et al., 2009). The clinical manifestations of the disease are mainly characterized by nonsyndromic ID (cognitive impairment and delayed speech development); however, autism has also been reported. Most of the reported families were from the Middle East with particular overrepresentation in consanguineous communities ( Amin et al., 2022). To our knowledge, 21 different mutations in the TRAPPC9 gene have been reported in different patients, mostly in homozygous conditions in consanguine families from different origins. Three compound heterozygous mutations were also reported in the French, Thai, and Italian families. This study supported the previous findings and added a novel missense mutation to the genetic pool for autosomal recessive ID.
PLOD3 gene on chromosome 7q22 encodes lysyl hydroxylase 3 (LH3), which is involved in posttranslational modification of collagens. Pathogenic variants in PLOD3 lead to a deficiency in the enzyme LH3 causing a connective tissue disorder with a variable clinical presentation ranging from isolated contractures and bone fragility to a multi-system disorder. The features include joint contractures, scoliosis, low bone mineral density and fractures, sensorineural deafness, myopia, ocular abnormalities, rupture of a vascular aneurysm, skin and nail abnormalities, and dysmorphic features. There are very few cases reported in the literature ( Ewans et al., 2019).
The patient reported in this article had ID and developmental delay secondary to a homozygous variant in the TRAPPC9 gene as well as extensive contractures all over secondary to a homozygous variant in the PLOD3 gene. To the best of our knowledge, we are reporting for the first time the inherited syndrome having mutations in the two TRAPPC9 and PLOD3 genes leading to the disease. Any variation in the gene of PLOD3 leads to amino acid changes in the LH3 polypeptide chain localized in regions responsible for LH3 glycosyltransferase and lysyl hydroxylase activities. The significances of these mutations to LH3 enzyme activities and protein expression changes accordingly, leading to a phenotype change in which LH3 concentration is low and all LH3 activities, GT, LH, and GGT, are abnormally reduced and these changes contribute more commonly to connective tissue disorders.
The combination of homozygous mutations led to a complex neurodevelopmental disorder. She exhibited features of disorders caused by mutations in both genes, which is an extremely rare phenomenon not previously reported in the literature.
CONCLUSION
We reported the clinical and molecular characteristics of a young girl who had two novel homozygous variants in the PLOD3 and TRAPPC9 genes leading to bone fragility with contractures and intellectual developmental disorder. This report adds to the literature a unique case of a complex neurodevelopmental disorder caused by mutations in two different genes leading to a complex clinical phenotype. Consanguinity remains a major contributor to autosomal recessive disorders, and corrective actions are required to mitigate this risk. We suggest that screening of such families must be done by using WES to add new genes and novel mutations to the literature. Our study is a step forward, and it will help add to the literature on the disease in the Saudi population.