
Background
Sudden unexpected cardiac arrest is one of the interventions of the pre hospital emergency medicine teams. Sudden cardiac death (SCD) is described as a sudden death from cardiovascular origin occurring within 1 hour of symptom onset when witnessed, or when the individual was alive within the previous 24 hours when not witnessed [1]. Globally, SCD accounts for 4-5 million deaths per year and is in most cases linked to coronary artery disease [2]. Other causes include cardiomyopathies and channelopathies.
We present two unconscious patients with life-threatening cardiac arrest who were unsuccessfully resuscitated by the emergency team. As the cause of SCD was unclear an autopsy was performed.
Case Presentation Patient 1
The first case concerned a 44 years old Caucasian man who was found unconscious on the bed of his camper. Bystander cardio-pulmonary resuscitation (CPR) was immediately started and 15 minutes later the emergency team arrived. The patient was on asystole and had bilateral fixed mydriasis. Advanced life support (ALS) was started in accordance with the guidelines algorithm [3]. CPR was continued and the patient was transferred to the hospital. Unfortunately, there was no reaction and after 45 minutes in toto the patient was declared dead.
The patient had no previous complaints or symptoms at all. There was no illness, nausea, vomiting, headache, or other signs or symptoms. He was in follow-up for a possible familial history of sudden death. His last consultation dated 3 years ago and did not reveal any abnormalities.
Postmortem examination disclosed an enlarged heart (470 gr) with dilatation of both atria as well as the right ventricle. The presence of a “dimple” at the anterior side of the right ventricle was suggestive of dilatation. Microscopical investigation showed transmural interstitial fibrosis with associated reactive hyperplasia of persisting cardiomyocytes (Figure 1). There was no replacement fibrosis but rather interstitial perimyocyte-type fibrosis. Identical lesions were found septobasal although less pronounced. Perivascular fibrosis was present reaching into the trabeculae carneae as well as in the papillary muscles.
The right ventricle as well as the coronaries did not show any microscopical abnormalities.
Next-generation sequencing (NGS) disclosed a pathogenic mutation c.772G>A (p.Glu258Lys) in the myosin binding protein 3 (MYBPC3 gene) (transcript NM_000256.3) (Tables 1 and 2).
Case Presentation Patient 2
The second case concerned a 42 years old Caucasian man who was an obese active tobacco smoker and drug user [cannabis, cocaine, and sporadically MDMA (ecstasy)] with antecedents of hypertension. He was found unconscious by his mother. Bystander CPR was not started. When the emergency team arrived, the patient was in asystole and mydriasis nonreactive to light. There was no reaction despite ALS.
This patient had just been discharged from the hospital. He was admitted to the hospital for a sudden atypical thoracic pain syndrome. The patient described it as a sharp stabbing, movement and inhalation-related pain. He was feeling badly and weakly without vomiting, palpitations, dyspnea, or temperature.
Clinical examination was negative, and the blood results showed mildly impaired renal function and elevated D-dimers. Other investigations (ECG, chest X-ray, ventilation/perfusion scintigraphy were reassuring. During hospitalization, an elevated c-reactive protein (CRP) of 160 mg/l (normally <5 mg/l) and an elevated troponine I of 50,32 pg/ml (normally < 13pg/ml) were noticed. A transthoracic echocardiography showed some pericardial fluid, a borderline hypertrophic left ventricle, and a light to moderate dilatation (4,2 cm) of the proximal aorta above the aortic valve. The differential diagnosis of perimyocarditis and cocaine-induced myocarditis was made. The patient was treated with acetylsalicylic acid (Aspirin) with pain relief and normalization of CRP. Troponine I level remained high.
Postmortem examination showed an enlarged blue–red pericardial sac containing 562 gr clotted blood suggestive of hearttamponade. Inspection of the aorta ascendens revealed a dissection of 2/3 of the aortic wall reaching from the aortic arch down to the supravalvular region rupturing into the pericardial sac (Figure 2 & 3). Histological examination of the aortic wall showed mucoid degeneration (Figure 4).
Examination of the heart (581 gr) disclosed a firm left ventricular wall with a thickness of up to 15 mm anterolateral. Reactive hyperplasia of persisting cardiomyocytes was shown by microscopical investigation. There was no replacement fibrosis but rather interstitial perimyocyte type fibrosis. There were no indications for congenital heart disease nor for coronary artery disease. The aortic valve and the other cardiac valves were normal. Mild chronic lymhocytic-histiocytic pericarditis was confirmed. Serological investigations for both echo-and coxsackie viruses were negative. Toxicological screening was positive for cannabis and metabolites but negative for cocaine and amphetamines.
NGS disclosed a heterozygous c.1721G>A p. (Arg574Gln) missense variant in exon 18 of the MYBPC3 (transcript NM_000256.3). Additional heterozygous variants associated with the thoracic aortic dissection (TAD) were found such as c.1942G>T p.(Ala648Ser) in exon 27 of the elastine (ELN) gene ( transcript NM_ 173054.2) and c.4199G>C p.(Gly1400Ala) in exon 32 of the myosin 11heavy chain (MYH11) gene(transcript NM_001040114.1) (Tables 1 and 2).
| Arrythomogenic right ventricle cardiomyopathy: CTNNA3, DES, DSC2, DSG2, DSP, JUP, PKP2, PLN, RYR2, TGFB3, TMEM43 |
| Brugada syndrome: ABCC9, CACNA1C, CACNA2D1, CACNB2, GPD1L, LKCND3, KCNE3, KCNE5, KCNH2, KCNJ8, PKP2, SCN1OA, SCN1B, SCN2B, SCN3B, SCN4B, SCN5A, SEMA3A, TRPM4 |
| Catecolaminergic polymorphic ventricle tachycardy: CALM1, CALM2, CALM3, CASQ2, KCNE1, KCNJ2, RYR2, TRDN |
| Dilative cardiomyopathy: ABCC9, ACTC1, ACTN2, ANKRD1, BAG3, CRYAB, CSRP3, DES, DSG2, DSP, FLNC, LAMA4,LAMP2, LDB3, LMNA, MYBPC3, MYH6, MYH7, MYL2, MYPN, NEXN, PKP2, PLN, RBM20, SCN5A, SGCD, TAZ, TCAP, TMEM43, TMPO, TNNC1, TNNI3, TNNT2, TPMI, TXNRD2, VCL |
| Early repolarisation syndrome - Idiopathic ventricular fibrillation: ABCC9, CACNAIC, CACNA2D1, CACNB2, CALMI, CALM2, CALM3, KCND2, KCND3, KCNE5, KCNJ8, NKX2-5, SCN5A |
| Hypertrophic cardiomyopathy: ACTC1, ACTN2, ANKRD1, CALR3, CASQ2, CAV3, CRYAB, CSRP3, DES, FHL1, FLNC, GLA, JPH2, LAMP2, LDB3, MYBPC3, MYH6, MYH7, MYL2, MYL3, MYLK2, MYOZ2, MYPN, NEXN, PLN, PRKAG2, RYR2, TCAP, TNNC1, TNNI3, TNNT2, TPM1, TTR, VCL. |
| Long QT syndrome: AKAP9, ANK2, CACNA1C, CALM1, CALM2, CALM3, CAV3, KCNE1, KCNE2, KCNE3, KCNH2, KCNJ2, KCNJ5, KCNQ1, NOSIAP, SCN1B, SCN4B, SCN5A, SNTA1 |
| Thoracic aortic aneurysm/dissection ABL1, CTA, ARIH1, BGN, COL3A1, EFEMP2/FBLN4, ELN,EMILIN1, FBN1,FBN2, FLNA, FOXE3, HCN4,IPO8, LMOD1, LOX, LTBP3, MAT2A, MFAP5, MYH11, MYLK, NOTCH1, PLOD1, PMEPA1/TMEPA1, PRKG1, SKI, SLC2A10, SMAD2, SMAD3, SMAD4, SMAD6, TGFB2, TGFB3, TGFBR1, TGFBR2 |
| PATIENT\GENE | MYBPC3 NM_000256.3 | ELN NM_173054.2 | MYH11 NM_001040114.1 |
|---|---|---|---|
| Patient 1 | c.772G>A p.(Glu258Lys) Pathogenic mutation | Wild type | Wild type |
| Patient 2 | c.1721G>A p.(Arg574Gln) VUS | c.1942G>T p.(Ala648Ser) VUS | c.4199G>C p.(Gly1400Ala) VUS |
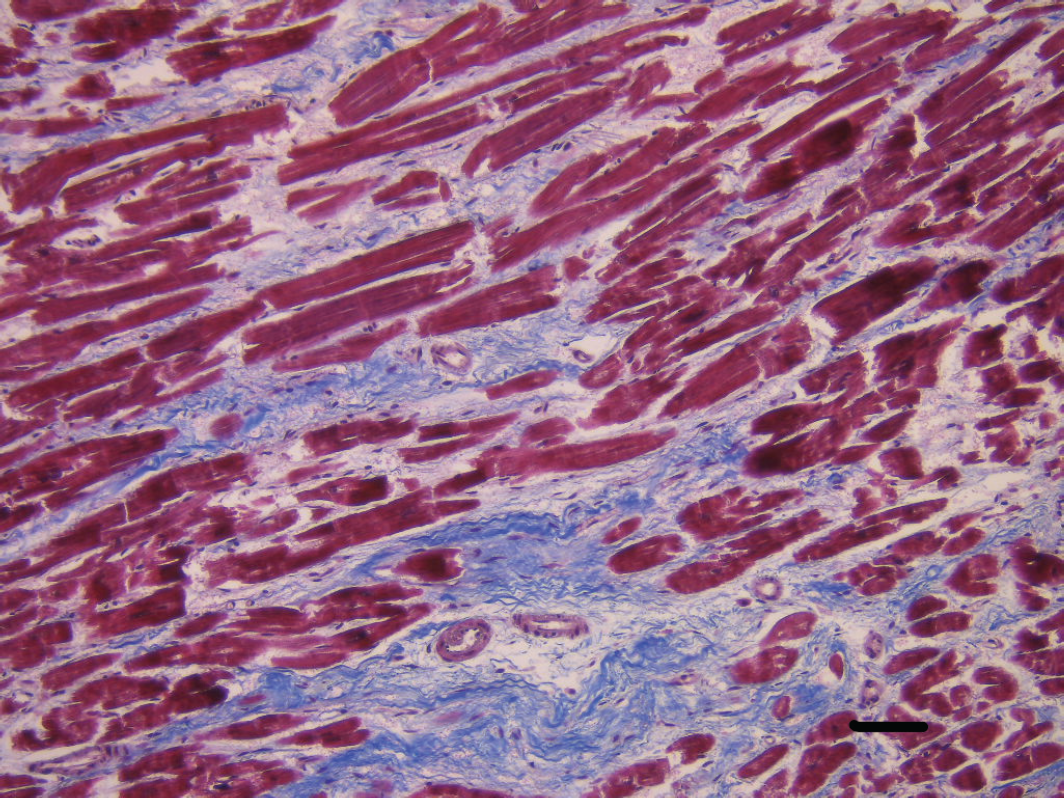
Dilative cardiomyopathy. Histopathology of the left ventricular wall displaying transmural interstitial fibrosis with associated reactive hyperplasia of persisting cardiomyocytes. No replacement fibrosis but rather interstitial perimyocyte type fibrosis with blue staining of the fibrotic connective tissue. (Masson’s Trichrome stain. magnification 200×. Scale bar: 50 µm).
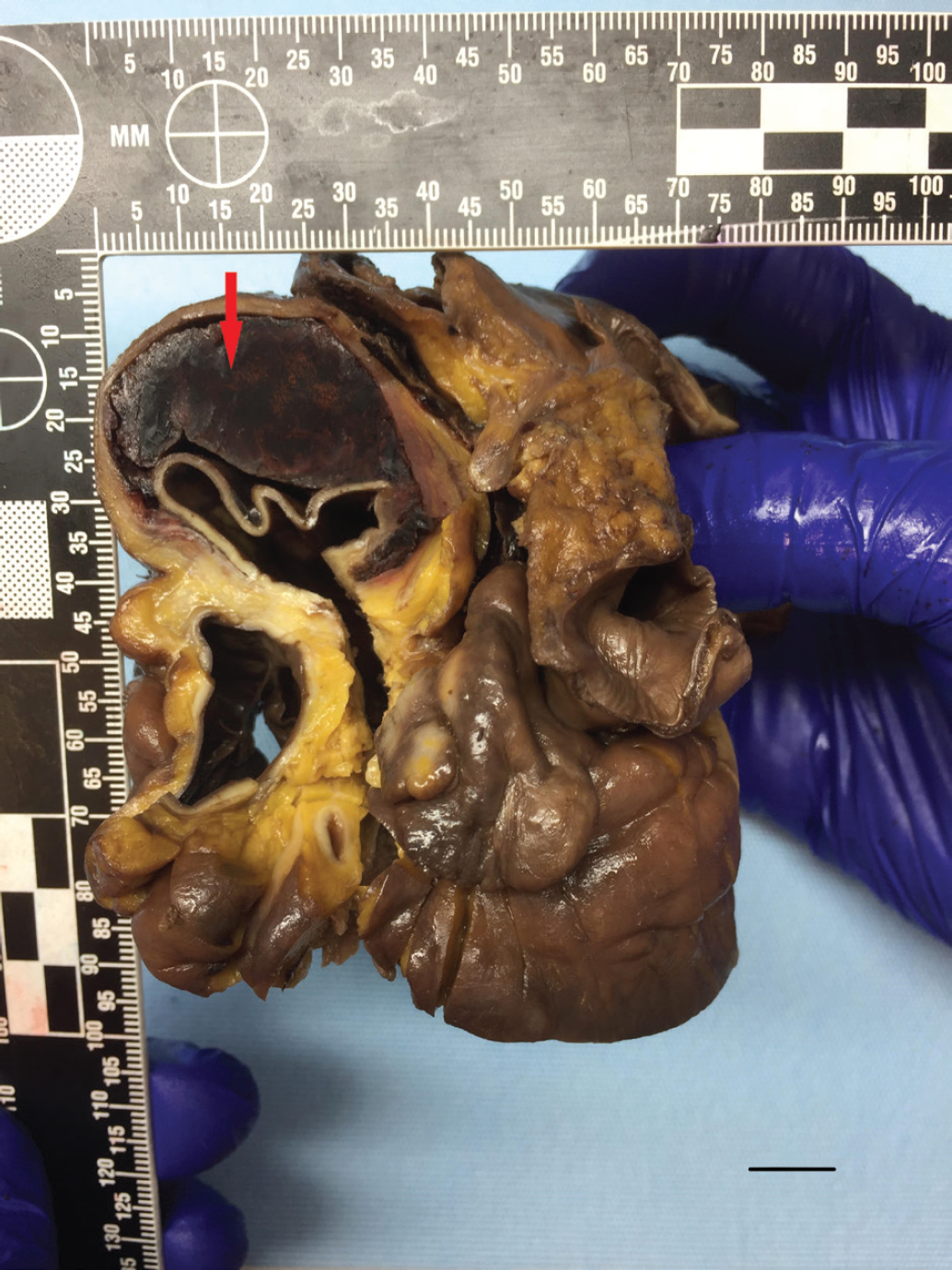
Macroscopy of the enlarged heart with transaxial opening of the ascending part of the thoracic aorta displaying dissection and clotted blood (red arrow). Scale bar: 1 cm.
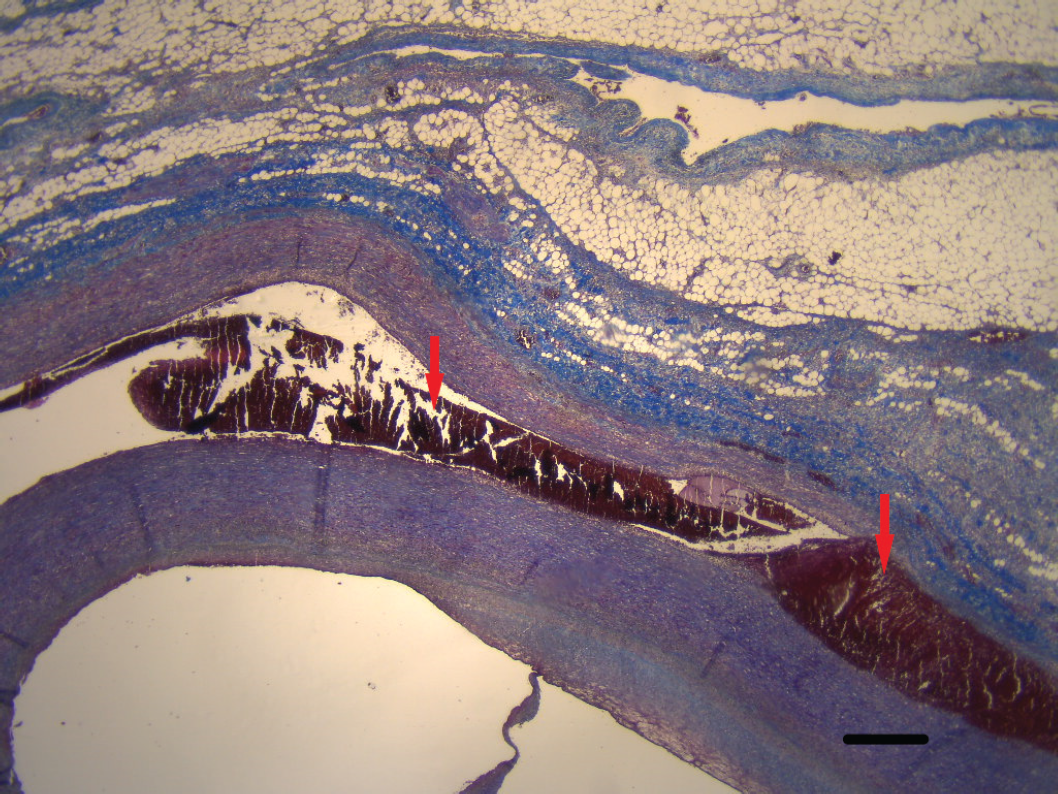
Microscopical view of the thoracic aorta with signs of dissection (red arrows) (Trichrome Masson connective tissue stain 20×. Scale bar: 500 µm).
All these variants were classified according to the standards and guidelines for the interpretation of sequence variants as recommended by the American college of medical genetics and genomics and the association for molecular pathology [4].
Discussion
Cardiomyopathies are a very heterogenous group of diseases including hyperthrophic cardiomyopathy, dilative cardiomyopathy, restrictive cardiomyopathy, non-compaction cardiomyopathy, and arrythmogenic cardiomyopathy. They all display structural abnormalities in the cardiac muscle leading to progressive loss of electrical stability and less effective systolic and diastolic function resulting in cardiac failure. Approximately 75% of patients with hypertrophic cardiomyopathy harbor mutations in genes encoding the thick filament components myosin heavy chain and myosin binding protein C (MYH7 and MYBPC3) suggesting that hypertrophic cardiomyopathy is a disease of the sarcomere [5]. In contrast, dilative cardiomyopathy is a more genetically heterogeneous disease including structural aberrations in genes coding for cytoskeletal, nucleoskeletal, mitochondrial, and calcium processing proteins [6].
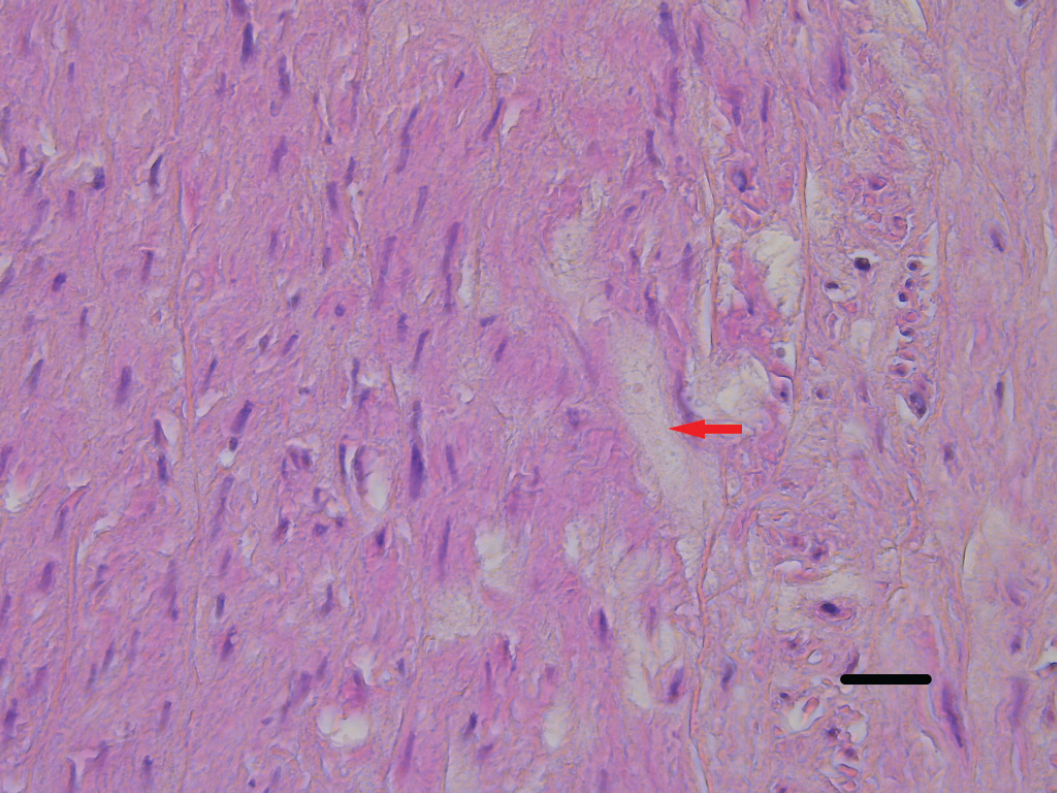
Degenerative mucoid change in the vessel wall of the aorta (red arrow) (Hematoxylin & eosin stain 400×. Scale bar: 25 µm).
In both patients, the autopsy disclosed macroscopical and microscopical features of dilative cardiomyopathy. The elevated Troponin I (50,32 pg/ml: normal value <13 pg/ml) found in our second patient can be due to this cardiomyopathy [7]. This raises the question of whether the drug abuse [cannabis, cocaine, and sporadically MDMA (ecstasy)] has induced cardiomyopathy [8-10]. Cannabis abuse can lead to an associated cannabis-induced stress cardiomyopathy [11]. The symptoms of chest pain, shortness of breath, and dizziness are highly suggestive. However, also chronic use of cocaine and ecstasy can induce dilative cardiomyopathy [9,10].
In both cases, we found heterozygous variants in the MYBPC3 gene. This gene codes for Myosin binding protein C playing a pivotal role in cardiac muscle contraction. Structural aberrations in this gene are associated with the development of dilative cardiomyopathy, hypertrophic cardiomyopathy, and non-compaction cardiomyopathy [5,6]. Our first patient showed a pathogenic mutation c.772G>A (p.Glu258Lys) in the MYBPC3 gene resulting in alternative splicing with deletion of exon 6. This defective protein leads to cardiac failure and subsequent SCD. Our second patient had a heterozygous c.1721G>A p. (Arg574Gln) missense variant in exon 18 of the MYBPC3, classified as a variant of unknown significance (VUS). This variant has been found in one patient with cardiomyopathy. The variant is described in the ClinVar databank and the Leiden open variation database databank as a VUS. NGS disclosed additional heterozygous variants associated with the development of TAD such as c.1942G>T p.(Ala648Ser) in exon 27 of the ELN gene and c.4199G>C p.(Gly1400Ala) in exon 32 of the MYH11 gene.
The second patient died as a consequence of this TAD with a rupture in the pericardial sac. The genetic profile may have played a role. However, hypertension and to a lesser extent cocaine abuse are also well-known risk factors for the development of thoracic aortic aneurysms [12]. The International registry of acute aortic dissections study suggests that cocaine is not likely to be responsible for more than 1% of aortic dissections but cocaine seems to play a significant role in precipitating aortic dissection among the young cohort in the study [13]. This raises the question of whether variants of unknown significance (VUS) are playing a role in the development of cardiomyopathy and aortic dissection. Pottinger et al. [14] described pathogenic or likely pathogenic variants in a small proportion of biobank subjects. The authors focused on 385 subjects known to harbor a VUS. They found a significant overall association between VUS and longitudinal changes in cardiac chamber dimensions. VUS seems to contribute to myocardial dysfunction and at least some VUS seem to be erroneously classified [15]. These findings of both dilative cardiomyopathy and thoracic aorta dissection could explain the fatal outcome in our second patient. The VUS found in our patient together with the other precipitating factors such as hypertension and drug abuse is known to play a role in the development of both dilative cardiomyopathy and TAD. Cocaine use is associated with several cardiovascular complications and SCD. Recent cocaine use leads to an increased risk for TAD. Abuse of cocaine can be associated with the development of both TAD as well as cardiomyopathy [9,12,16].
These findings suggest that it may prove very useful to perform an autopsy in every case of unexpected SCD and to combine this with adequate genetic testing. Genetic testing can give more insight into how a specific pathology develops in a patient. In our first patient, we found a pathogenic mutation in the MYBPC3 gene resulting in alternative splicing with deletion of exon 6. The resulting aberrant protein structure gave rise to myocardial dysfunction resulting in sudden death. The situation is less straightforward for our second patient. We found several VUS in both genes coding for structural myocardial contractile proteins (MYBPC3) as well as in proteins associated with the stability and integrity of the cardio-vascular structures (ELN and MYH11). It is most likely that these VUS in combination with other precipitating factors such as hypertension and drug abuse led to the development of cardiomyopathy and TAD resulting in fatal outcome.
If the cause of SCD is unclear, an autopsy is mandatory, especially the so-called “molecular” autopsy. Genetic screening can be beneficial to detect genetic susceptibility to dysrhythmias (long QT syndrome, Brugada syndrome), cardiomyopathies, thoracic aortic aneurysms, and dissection. Genetic analysis closely correlated with the postmortem findings and significant clinical information can be used to decide whether genetic screening and clinical follow up of relatives should be carried out as in both described patients.
Conclusion
SCD can be the final stage of several pathologies. In acute myocardial infarction due to coronary artery disease the underlying cause is clear. The situation is however more complex in drug abuse where the physician should be aware that underlying pathologies can be masked. If the cause of sudden cardiac arrest is unclear, a “molecular” autopsy is recommended to detect genetic susceptibility to dysrhythmias (long QT syndrome, Brugada syndrome), cardiomyopathies, thoracic aortic aneurysms, and dissection. Correlating significant clinical information, postmortem findings, and genetic analysis can be used to detect underlying pathologies and to decide whether genetic screening and clinical follow up of relatives should be carried out [2]. It is mandatory to interpret these findings together with the clinical information. However, more research is warranted to address the role of certain variants of unknown significance in the development of cardiovascular disease and their usefulness in assessing the risk for relatives.