
Background
Despite the real progress in contemporary medicine, the pathology of the right heart continues to represent a great challenge for physicians. For a long period, the right ventricle was considered as a passive chamber of the heart with no clear clinical importance in the patient’s prognosis. Pulmonary arterial hypertension (PAH) represents a nosological entity, which may involve multiple clinical conditions and can complicate many respiratory and cardiovascular diseases. Furthermore, PAH may be idiopathic with severe impact on the right ventricular (RV) function and structure and poor outcome of the patient [1]. Cardiac magnetic resonance (CMR) is an non-invasive “gold standard” for the assessment of the right heart structure and function [2] and, together with right heart catheterization (RHC), may offer important information about different causes of the right heart pathology [1]. We hereby present an interesting and challenging case of PAH due to partial anomalous pulmonary venous return in a young woman.
Case Presentation
A 41-year-old Caucasian woman, with no past medical history of cardiovascular or any other pathology and with no regular medication presented to a cardiologist with the complaints of the shortness of breath during moderate physical activity. The patient mentioned that symptoms occurred approximately 2 months ago and evaluated progressively until present. Before that period, the patient was extremely active, practiced physical activity every day. At physical examination, she had a regular pulse (102 b.p.m.), with no heart murmurs and no distension of neck veins; her blood pressure was 110/70 mmHg; the patient’s ankles were a little swollen. Lung auscultation did not reveal any pathological signs. Respiratory rate was 17 b.p.m. with an oxygen saturation of 95%.
When a detailed medical history was collected, the patient mentioned that she had traveled by plane for the first time in her life for a short holiday and had a traumatism of the right ankle about 2½ months ago. There were no history of syncope and no relatives with premature death.
Electrocardiogram (ECG) showed inverted T-waves in V1-V5 and moderate right axis deviation with no signs of complete right bundle-branch block (Figure 1).
Transthoracic echocardiography revealed severe dilatation of right heart chambers, RV pressure, and volume overload with no obvious data for intra-cardiac shunts, moderate-to-severe tricuspid regurgitation, and pulmonary artery systolic pressure (sPAP) of 84 mmHg. The RV function was deteriorated: tricuspid annular plane systolic excursion of 14 mm, s’ 0.08 m/s, and fractional area change 29% with no signs of regional RV wall abnormalities. Furthermore, we mentioned the dilatation of the RV outflow tract (RVOT) (parasternal long axis view 37 mm, parasternal short axis view 39 mm) and pulmonary artery dilatation (32 mm). The patient was redirected for the admission to the Institute of Cardiology (third-level hospital) with the possible diagnosis of chronic thromboembolic PAH. The laboratory results included D-Dimer level of 246.00 ng/ml (ref. range: 0-500 ng/ml), cardiac troponin I level < 0.50 ng/ml (ref. range: 0-0.5 ng/ml), and NT-proBNP level of 535.00 ng/ml (ref. range: 0-300 ng/ml). At the hospital, the patient was also consulted by a rheumatologist who recommended additional laboratory examinations that were shortly performed. These analyses included anti-dsDNA immunoglobulin (Ig) G 8.4 U/ml (ref. range < 25 U/mL), negative anti-MPO IgG and anti-PR3 IgG, homocysteine 9.63 μmol/L (ref. range ≤12), positive lupus anticoagulant, anticardiolipin antibodies: IgG < 2 GPL/ml (ref. range < 20 GPL/mL) and IgM 10.3 MPL/ml (Ref. range <13), antibeta-2 glycoprotein 1 IgG 3.2, IgM 4.2, and IgA 3.5 U/ml (ref. range <5), erythrocyte sedimentation rate of 2 mm/ hour (ref. range 2-15 mm/hour), serum iron of 9.2 μmol/l (ref. range 6.6-25.9 μmol/l), and C-reactive protein of 2.69 mg/l (ref. range < 5). Furthermore, we performed a capillaroscopy that did not reveal any pathological changes in small vessels of the microcirculation in the nail fold.
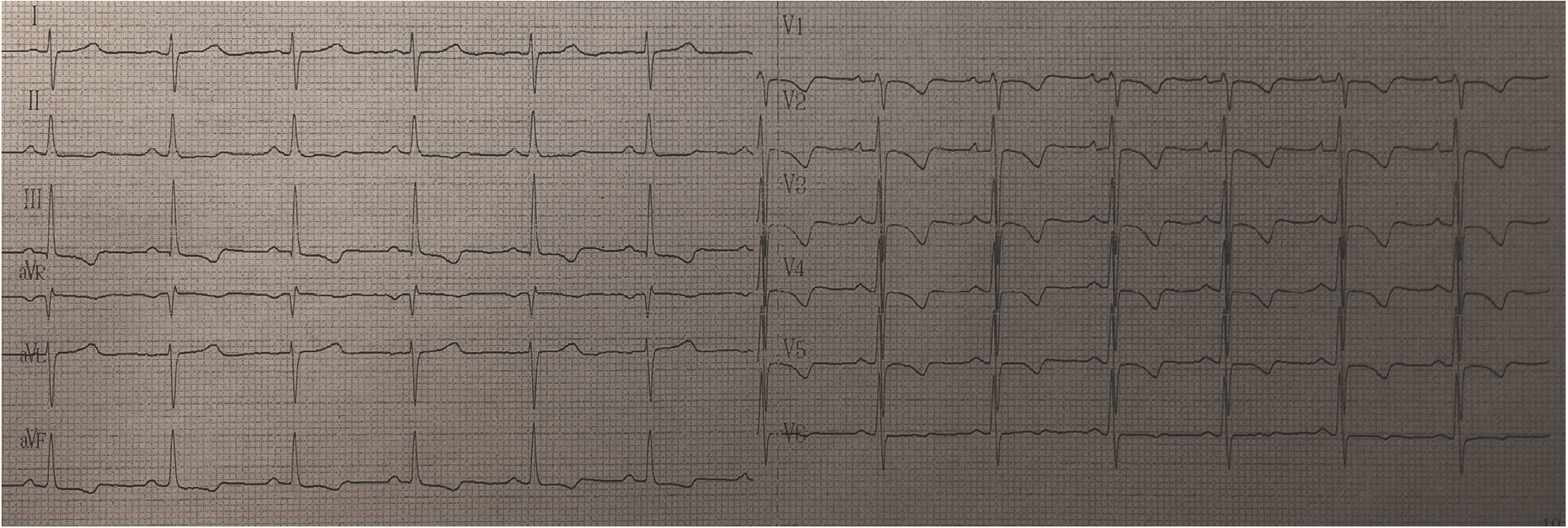
ECG of the patient. Inverted T-waves in V1-V5 and moderate right axis deviation with no signs of complete right bundle-branch block.

Computed pulmonary tomography angiogram. (A, B, and C) Axial C + arterial phase, showing normal aspect of pulmonary arteries. (D) Coronal view: no pathological changes suggestive for pulmonary artery embolism were revealed.
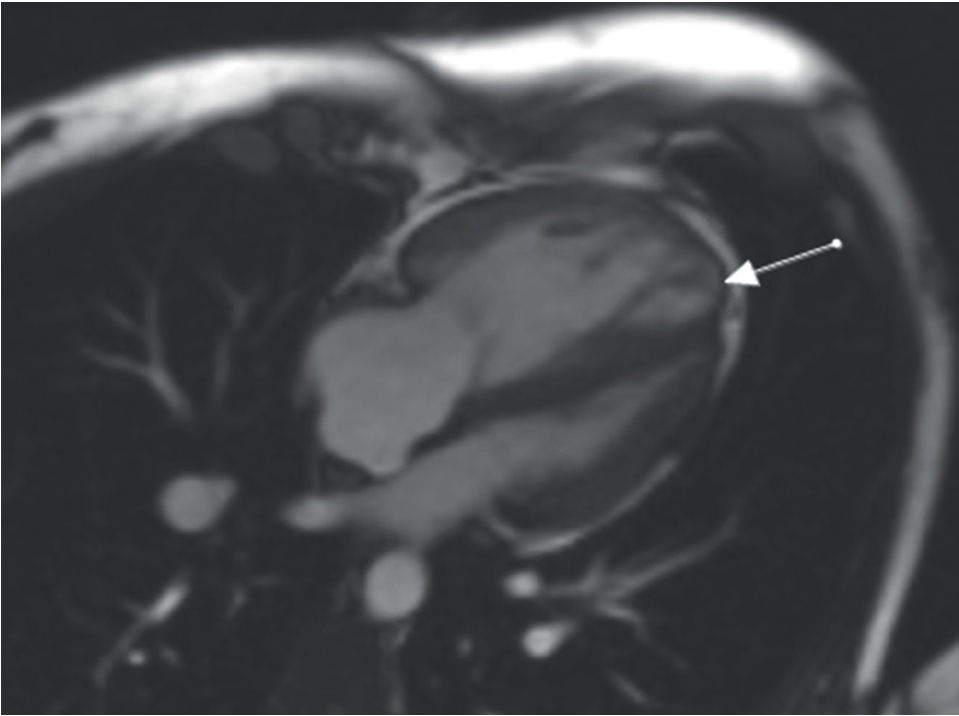
CMR. Long axis four-chamber view cine images show the signs of dyskinesia in the anterior wall in the trabeculated myocardium of the right ventricle.
Computed tomography (CT) pulmonary angiography was performed with no imaging evidence of suspected pulmonary artery embolism (Figure 2).
On CMR, the RV was severely dilated compared to the left ventricle (LV): DTD of RV 44 mm, RVOT 33 mm, and Volume telediastolic (VTD)/Body surface area (BSA) 107 ml/m2. The ejection fraction of RV was 22% (ref. range: 47%-74%). The thickness of RV myocardium was 2-3 mm in diastole. The signs of dyskinesia in the anterior wall in the trabeculated part of RV were present (Figure 3). More of that, late gadolinium enhancement (LGE) was present in the inferior septal segment of LV near RV (secondary sign of pulmonary hypertension) and the anterior septal basal segment of LV of non-ischemic etiology (Figure 4A). Furthermore, we mentioned pronouncedly expressed LGE in the epicardium of RV of non-ischemic etiology (Figure 4B). The epicardial adipose tissue of 5-6 mm was detected on the anterior wall of RV and 3-4 mm on the lateral wall of LV (Figure 5). EF of LV was 49%, with DTD of 41 mm. According to the obtained CMR data, we suspected the diagnosis of arrhythmogenic RV cardiomyopathy [3–5].
Holter ECG monitoring 24 hour showed solitary premature ventricular and supraventricular contractions with no other evidence for arrhythmias.
RHC confirmed the diagnosis of PAH with mean pulmonary arterial pressure of 54 mmHg and pulmonary arterial wedge pressure of 14 mmHg. However, we observed one more interesting thing-a step up in oxygen saturation from 68% at the high superior vena cava (SVC) to 83% at the level of the right atrium with a calculated pulmonary-to-systemic flow ratio (Qp/Qs) of 1.7. The repeated CMR showed a partial anomalous of pulmonary venous return (PAPVR) from the right upper and medium lobes to the mid-SVC (Figure 6).

CMR. (A) short axis two-chamber view: LGE present in the inferior septal segment of LV near RV; (B) anterior septal basal segment of LV of non-ischemic etiology.

CMR. Epicardial fat of 5-6 mm detected on the anterior wall of RV (white arrows) and 3-4 mm on the lateral wall of LV (yellow arrow).
The patient was started on tadalafil 20 mg PO daily and torasemide 5 mg PO daily. She also was referred for surgical repair, but due to personal considerations and good clinical condition on medical therapy, she refused to undergo the proposed surgery and elected close monitoring on medical treatment.
During outpatient follow-up 1 month after discharge, clinical improvement was substantial (the 6-minute walk distance > 440 m) with a better quality of life. Transthoracic echocardiogram showed a lower sPAP of 59 mmHg.
Discussion
Partial anomalous pulmonary venous return is a rare grown-up congenital heart disease, which could result in a significant left-to-right shunt and PAH or remain asymptomatic for a long period of time [6,7]. First, it was described by Winslow [8]. The retrospective study, based on chest CT reports, revealed PAPVR only in about 0.1% images [9]. Clinical manifestations can occur at any age. The most common clinical condition is a progressive dyspnea. The first diagnostical tool is transthoracic echocardiography, but it lacks the possibility of good three-dimensional (3D) visualization of pulmonary veins and their relationship to the left atrium [10]. Echocardiography concentrates the attention on the involvement of the right heart, and we should find the etiology of PAH. CT angiography and CMR angiography both are excellent imaging modalities for anomalous pulmonary veins [10], but sometimes, as we report, the main clue to the correct diagnosis is a step up in oxygenation during RHC [7]. For the correct therapeutic decision-making, shunt fraction calculation is recommended. If in a symptomatic patient with right heart enlargement, the significant left-to-right is present (Qp/Qs > 1.5), surgical repair is recommended [6,7]. Surgical outcomes for PAPVR are usually with a low complication rate [6,7]. For those patients who refuse surgical repair or have severe concomitant pathology, the treatment with pulmonary vasodilators could represent a temporary bridge to the surgery [1,11].
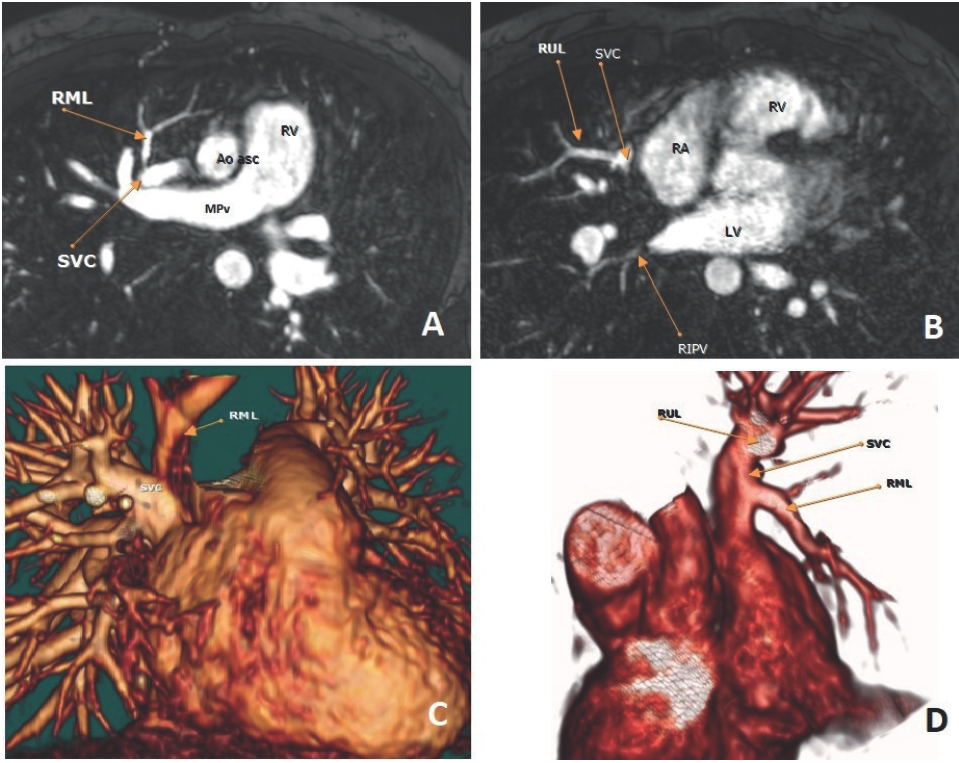
CMR. PAPVR from the right upper and medium lobes to the mid SVC. (A) Axial plane magnetic resonance imaging (MRI) angiographic image shows the right middle lobe accessory pulmonary vein drainage into SVC; (B) the same plane angiographic image present right upper lobe accessory pulmonary vein attached to SVC; (C and D) 3D virtual reality MRI image shows the right middle lob and right upper lobe accessory pulmonary veins attached to SVC.
Conclusions
PAPVR is a rare congenital heart disease that could be silent for a long period of time and represents a real diagnostical challenge for physicians. The main symptom is dyspnea and the development of PAH. Step up in oxygen saturation during RHC is crucial in the diagnosis of PAPVR, which is a potentially curable condition. Early surgical repair should be preferred in symptomatic patients.
What is new?
Pulmonary arterial hypertension involves many different clinical conditions and has an important impact on RV function and patient’s prognosis. Some of these conditions can be potentially curable, for example, congenital heart disease that could be surgically repaired. The authors present an interesting and challenging case of PAH due to partial anomalous pulmonary venous return in a young woman.