
Background
Idiopathic normal pressure hydrocephalus (iNPH) is usually regarded as a disease characterized by gait and balance disturbance, cognitive dysfunction, and urinary symptoms caused by a disturbance of the cerebrospinal fluid (CSF) dynamics [1]. The diagnosis of probable iNPH is based on clinical features, brain imaging, and CSF dynamics [2]. However, the pathophysiology of iNPH is not fully explained. In the last few years, there is an increasing number of familial iNPH, which raises the suspicion of a genetic component [3]. Recently, Korhonen et al. [4] presented the prevalence of chromosome 9 open reading frame 72 (C9ORF72) expansion among Finnish iNPH patients. In their study, 487 patients with possible iNPH diagnosis were controlled and 8 patients (1.6%) were carriers of C9ORF72 expansion. In the same study, there was one patient who was diagnosed with amyotrophic lateral sclerosis (ALS), and in all expansion carriers, the family history was positive for either ALS, iNPH, dementia, psychiatric diseases, gait problems, or a combination of all above mentioned diseases. The C9ORF72 expansion is known to cause frontotemporal lobar degeneration and ALS [5,6]. We report an interesting case of a patient with probable iNPH who also developed ALS.
Case Presentation
A 64-year-old Caucasian woman, with hypertension since the age of 35, hypothyroidism, family-free history for iNPH, dementia, psychiatric disease, ALS, and gait disorders, was referred to our emergency room, the university hospital in Linköping, because of a generalized tonic-clonic seizure. She experienced Todd’s paresis on her left side, including left facial paresis. Neurological assessment using the National Health Institute Stroke Scale (NIHSS) showed three points for left central facial paresis, one point for left complete hemianopia, and one point for partial oculomotor nerve paresis. A brain computed tomography (CT) revealed an obvious ventriculomegaly disproportionate to cerebral atrophy, Evans index of 0.34, corpus callosum thinning and elevation with a callosal angle with 64o, widening of temporal horns (without hippocampal atrophy), widening of third ventricle at 20 mm, narrowing of Sulci and subarachnoid spaces over the high convexity and midline surface of the brain, and ballooning of frontal horns. The radiological investigation raised the suspicion of normal pressure hydrocephalus (NPH). A brain magnetic resonance imaging (MRI) showed aqueductal flow void, a normal width of the third ventricle, and no CSF obstructions. There were slight chronic ischemic changes in the periventricular areas, and the diffusion-weighted magnetic resonance imaging (DWI) associated with apparent diffusion coefficient (ADC) showed no infarction (Figure 1). The patient was admitted to our neurological department for observation. After 6 hours, a new assessment of the NIHSS showed 0 points and the patient had made a full recovery. An acute electroencephalogram (EEG) test showed suspect epileptic activity in the frontotemporal brain region. The electrocardiography test and telemetry were normal, the thyroid-stimulating hormone was 0.03 (0.4–4 mU/l), and T4 was at 21.4 (9–22 mU/l), all the other blood controls were normal. A sleep-deprived EEG test, compared to the previous EEG test, showed distinct improvement with some abnormal activity at the frontotemporal brain region and lack of epileptic activity. The patient was dismissed without any antiepileptic drugs, except for antihypertensive medication because of the continuous high blood pressure, and a follow-up was planned.
Despite our desire for immediate follow-up, the patient showed up to our outpatient clinic 4 years later because of balance problems. Cognitive impairment and urinary problems were lacking. The neurological examination showed distinct impaired vibration in both feet. However, she decided to be dismissed, despite our recommendation for polyneuropathy investigation.
One and a half years later, after an upper respiratory infection, her balance and bladder control were impaired. A new brain MRI, compared to the previous one, demonstrated an increased Evans index of 0.37 and the dimension of third ventricle was 12 mm. There were no microangiopathy changes in the periventricular white matter. Radiological imaging fulfilled the criteria for iNPH diagnosis.
During the period when iNPH was investigated, the patient’s balance became insidiously impaired and the gait could be characterized as being magnetic and broad with some sensory deficits from the lower limbs. Romberg’s test was impaired. The start–stop test, reflexes, limp-power test, and ataxia test were normal under neurological investigation. The gait test was compatible with the iNPH disease with decreased step height and length, decreased cadence, increased trunk sway during walking, turned-out toes on walking, widened standing base, turning bloc, and retropulsion. Motor evaluation showed 15 sec for the timed up and go (TUG) test, 23 steps for TUG step, 12 seconds for 10-m walk time test (w10mt), and 23 steps for 10-m walk step test (w10ms). Cognitive evaluation showed a mini-mental statement (MMSE) test with 26 points, slow psychomotor, and executive ability. A lumbar puncture (LP) was carried out with the patient lying in the lateral decubitus position, and the opening pressure was 20-cm H2O followed by CSF drainage of 50 ml. There was motor impairment after the tap test with a TUGt 13 sec (–8%), TUGs 24 steps (–4%), w10mt 13 sec (+13%), and w10ms 24 (–4%). The patient responded negatively to the tap test. The neurodegenerative markers showed a tau of 147 ng/l (normal <400 ng/l), β-amyloid of 420 ng/l (normal >550 ng/l), and f-tau of 19 ng/l (normal <80 ng/l). No data for neurofilaments (NFL) were available.
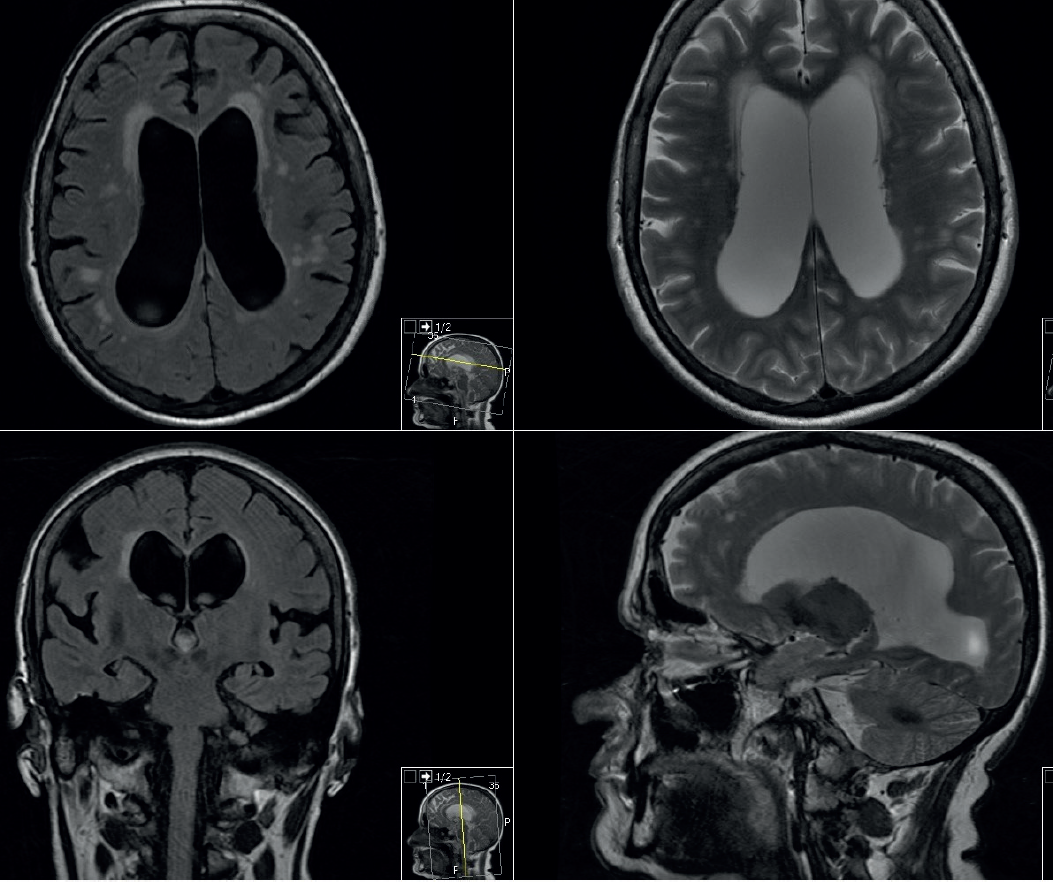
Brain MRI demonstrated aqueductal flow void and a normal width of the third ventricle. DWI associated with ADC showed no infarction.
Three months later, the patient came for a follow-up and she described impairment of iNPH symptoms. A new motor evaluation, compared with the results from the first control, showed w10mt 18 seconds (–50%), w10ms 31 steps (–35%), TUGt 24 seconds (–60%), and a TUGs 34 steps (–48%). The MMSE was unchanged. The radiologic picture in combination with the symptoms and signs compatible with iNPH led us to accept her for a ventriculoperitoneal shunt operation, and she had improved significantly at the 3-months follow-up visit.
Nine months after the shunt operation, the patient complained of bilateral leg weakness and the possibility of a dysfunction of the shunt was considered. The brain CT and shunt overview were normal. The neurological evaluation showed bilateral drop foot, minor distal leg weakness and muscle atrophy with discreet diminished vibration in both feet, and a suspected sensorimotor polyneuropathy. The thorax and abdominal CTs with contrast agents were without malignancy and MRI of the whole spinal cord showed spinal stenosis at C5–C6 without myelopathy (Figure 2). Electroneurography (ENG) showed decreased amplitude, prolonged distal motor latency, and slowed conduction velocity consistent with axon loss. Needle Electromyography (EMG) revealed the presence of fasciculation and fibrillation potentials in all extremities. The results were compatible with motor neuron disease (NMD). A new LP showed lack of pleocytosis, normal albumin, and neurodegenerative markers with high tau at 1,030, normal β-amyloid at 1,080, slightly high f-tau at 88, and elevated NFL at 17,600. All paraneoplasmatic antibodies in the serum were negative. A muscle biopsy showed pronounced pathological changes with atrophic fibers and myopathy-related changes secondary to neurological disease. The clinical, neurophysiological, and CSF evaluations led to an ALS diagnosis. The patient’s clinical status decreased rapidly with the involvement of bulbar and respiratory symptoms, and she died one year after the diagnosis of ALS and 6 years after she first visited our department.

Discussion
iNPH is a disease of the elderly population [7]. Our patient had ventriculomegaly at the age of 64, but the balance and gait disturbances appeared at the age of 68. In order to be diagnosed, a patient with iNPH needs a combination of clinical symptoms, such as gait disturbance, balance impairment, cognitive decline, and urinary disturbance [8]. Our patient had an epileptic seizure which probably was unrelated to her later disease, although it has been previously described as a possible symptom of iNPH in the literature studies [9,10]. According to the iNPH Radscale, our patient fulfilled the radiological criteria for iNPH [11]. There was no abnormality of CSF neuro-degenerative markers and no known cause of secondary NPH was found. There was a period of 4 years between the first brain CT and preoperative brain MRI and there was a radiological progress in ventriculomegaly. The clinical evaluation was not compatible with any other neu-rodegenerative disease. During the iNPH investigation, the patient experienced motor and cognitive impairments which met the international criteria for probable iNPH [12]. At the 3-month follow-up, there was a clear improvement of the patient’s symptoms. Therefore, we conclude that our patient suffered from iNPH.
A short period after the shunt operation, the patient’s motor condition impaired and presented clinical symptoms which raised the suspicion of NMD. The evidence of lower motor neuron degeneration by clinical, electro-physiological, neuropathological examinations in combination with high NFL led to the diagnosis [13]. Hence, the diagnosis of ALS was confirmed due to the progressive symptomatology.
In Korhonen et al.’s study [4] group of 409 iNPH patients, 324 were shunt responders and 85 nonresponders. The prevalence of the C9ORF72 expansion was detected in both groups (six patients in the responder’s group and 1 patient in the nonresponder’s group). The mean age at the onset of the symptoms was lowered compared to non-carriers (59 versus 70 years). Our patient appeared with tetra-ventriculomegaly at the age of 64 and developed gait disorders at the age of 68. Korhonen et al.’s patient, who was a carrier of the C9ORF72 expansion, was diagnosed with iNPH at the age of 52, was operated with a shunt at the age of 55, was reevaluated at the age of 70 because of dysfunction, and was diagnosed with ALS. Response to the shunt plays a significant role in confirming iNPH diagnosis. In our case, the patient was a shunt responder and fulfilled the international criteria for iNPH diagnosis. In contrast to Korhonen et al.’s patient, our patient lacked family history for iNPH, dementia, psychiatric disease, ALS, and gait disorders. One might think that there was a difference between these two cases because Korhonen et al.’s patient developed ALS 15 years after a shunt operation, while our patient had a more aggressive onset. There might be some other factors required along with this expansion for developing ALS. It would have been very interesting to make a genetic analysis to see if she carried the C9ORF72 expansion, but unfortunately this was not done.