
Background
Hypomyelination with brainstem and spinal cord abnormalities and leg spasticity (HBSL, OMIM 615281), described first as a severe infantile onset inherited white matter disorder affecting both the brain and spinal cord, is caused by autosomal recessive mutations in the DARS gene which encodes the cytoplasmic aspartyl-tRNA synthetase[1], an aminoacyl-tRNA synthetase (ARS). ARSs are essential enzymes responsible for attaching amino acids to their cognate tRNA molecules in the cytoplasm and mitochondria ensuring translational fidelity. Mutations in cytosolic and some mitochondrial ARS cause central and peripheral nervous system conditions[2–8].
The initial patient cohort with DARS mutations had progressive leg spasticity and never achieved independent ambulation [1]. DARS associated leukoencephalopathy can mimic steroid responsive neuro-inflammatory disorder as two cases presented in early adulthood with subacute spastic paraplegia had partial improvement with steroids[9]. These two cases demonstrated only minor focal hyperintense T2 signals supratentorially but identical spinal cord changes in the dorsal columns and corticospinal tracts as those with earlier presentation.
HBSL phenotypically resembles leukoencephalopathy with brainstem and spinal cord involvement and elevated lactate (LBSL). LBSL is caused by mutations in DARS2, which encodes the mitochondrial portion of aspartyl-tRNA synthetase[10]. These two conditions may share a common underlying molecular pathology[11]. Both DARS and DARS2 proteins are essential for development as knock-out mice die in utero[11,12]. Heterozygous Dars+/- mice display 50% of normal mRNA expression in the central nervous system but have an 80% reduction of DARS protein. Despite this Dars+/- mice were phenotypically normal[11] suggesting that a small amount of protein produces substantial impact on phenotype in HBSL. Pre-pulse inhibition, a measure of attentional processing, was, however, significantly reduced in Dars+/- mice[11].
LBSL is more heterogenous, from rapidly fatal infantile onset to mild slowly progressive adult onset. The commonest phenotype of LBSL is childhood onset slowly progressive deterioration. The commonest form of HBSL, however, was severe infantile form.
Case Presentation
The proband presented at 3-year old with unsteady gait and frequent falls. Her parents are non-consanguineous and there was no significant perinatal history except for mild maternal hypertension and a diagnosis of left-sided unfused lumbar hemivertebrae and butterfly vertebrae following abnormal antenatal scans. She has bitemporal narrowing, plagiocephaly, short wide neck, epicanthus inversus, and over-folded right ear helix. She had mildly impaired early motor development, walking unsupported at 21 months. Examination revealed bilateral brisk knee reflexes, sustained ankle clonus and extensor plantar responses. A 1.5 Tesla magnetic resonance imaging (MRI) brain was performed at presentation showing congenital spinal abnormality of multiple hemi- and butterfly vertebrae but also extensive white matter signal abnormalities involving the peripheral white matter of both the cerebral hemispheres and the posterior columns of the spinal cord. MR spectroscopy demonstrated possible mild elevation of choline and N-acetylaspartate (NAA) compared to creatinine. Vitamin B12 deficiency was excluded. She was reviewed annually.
The examination at 12-year old revealed acquired microcephaly. She continued to fall frequently, complained of leg weakness and stiffness, and displayed an in-toeing gait bilaterally. Her examination showed upper motor neuron signs in the lower limbs along with cerebellar signs. She was also diagnosed with mild learning disability.
Sibling 1 is the proband’s younger brother. He walked independently at 2-year old. He was clumsy, had behavioral difficulties and poor concentration from 4-year old. He had brisk reflexes with extensor plantar responses and had a Chiari 1 malformation with craniocervical stenosis without a syrinx. Craniocervical decompression was performed at 7 years of age when he worsened with frequent falls, increasing headaches and deterioration in behavior and school work. He developed meningitis and post-meningitic hydrocephalus requiring a ventriculo-peritoneal shunt soon after the decompression. Since then, he has deteriorated in motor abilities, becoming clumsier. Although he is still walking independently, his memory has deteriorated, and behavior has become erratic. At 12-year old, he has a slight in-toeing mildly ataxic gait bilaterally, some cerebellar signs, brisk leg reflexes, and extensor plantar responses.
Sibling 2, the older sister of the proband, is the least affected. Early development and cognition were normal. She walked unsupported at 9-month old. Parents have noted occasional unprovoked falls. At 7 years, reflexes and tone were normal but plantar response were extensor bilaterally. At 14-year old, she had brisk upper limbs and lower limb reflexes, very mild in-toeing, ataxic gait and other cerebellar signs of dysmetria, and dysdiadochokinesia. Her academic progress and growth parameters were normal.
The siblings had gait analysis (video kinematic and kinetic analysis) performed. All three siblings displayed similar results of lacking full knee extension on walking with evidence of anterior pelvic tilt and a very mild internal foot progression. They were deemed to have functional gaits with no indication for surgical intervention. Clinical and radiological characteristics of the siblings were tabulated (Table 1 and Figure 1).
The proband was enrolled into the Deciphering Developmental Disorders (DDD) study [13]. Trio-based exome sequencing (Agilent SureSelect 55MB Exome Plus with Illumina HiSeq) and analysis as per Wright et al. [13] were performed. Array-based comparative genomic hybridization was also used to analyze copy number variation [Agilent 2 × 1M array comperative genomic hybridisation (Santa Clara, CA)]. Our proband has compound heterozygous mutations in the DARS gene: c.536G > A, p.(Arg-179Lys) (paternal allele) and c.1480C > G, p.(Arg494GLy) (maternal allele). The mutations identified were validated by Sanger sequencing and performed on her siblings. The c.536G > A, p.(Arg179Lys) mutation has not been reported. It lies slightly 5ʹ to all other reported mutations, but still within the 3’ Aminoacyl-tRNA synthetase class II domain. Although there is a relatively small physiochemical difference between the Arginine and Lysine amino acids, the Arg179 position is highly conserved and the p.Arg179Lys change is predicted to have a deleterious effect on protein function by the in silico analysis tools MutPred, Align GVGD, SIFT, and PolyPhen2.
Her siblings are also compound heterozygous for these mutations. Parental analysis confirmed the mutations to be in trans. The specificity of the associated phenotype, the pre-existing evidence to support the pathogenicity of the c.1,480C > G, p.(Arg494Gly) mutation, and the fact that all siblings are compound heterozygous for these two mutations represents very strong evidence to support the pathogenicity of both mutations.
Brain and spinal imaging findings were similar in the siblings with only minor variations (Table 1 and Figure 1).The proband and sibling 1 had high T2 and intermediate and low T1 signal in the centrum semiovale extending towards the white matter around the frontal horns and trigones of the lateral ventricles. This was less severe in sibling 2 as there was only minor patchy signal change in the centrum semiovale bilaterally along with the other MRI changes seen in her siblings.
This siblings’ MRI changes were generally milder when compared to other known cases of DARS related HBSL. Notably, there is no subcortical T1 hyperintense rim or abnormal signal in the internal capsule, no changes in the corpus callosum and no brainstem or cerebellum abnormalities. More recent brain and spine imaging for sibling 1 were post craniocervical decompression and meningitis making interpreting findings from DARS mutation difficult.
Discussion
Leg spasticity may have later onset in some symptomatic individuals with HBSL, thus not a reliable finding. We noted in-toeing gait in all siblings and propose this is due to subtle hypertonia in the internal rotators of the hip or the adductors and inverters of the foot secondary to the central nervous involvement of DARS mutation.
| PROBAND | SIBLING 1 | SIBLING 2 | |
|---|---|---|---|
| Patient characteristics | |||
| Gender | f | m | f |
| Pregnancy/delivery/perinatal period | Maternal hypertension, born at term, plagiocephaly, left sided L3/4 unfused hemivertebrae, butterfly vertebrae L1/2. | Maternal hypertension, born at term. | Maternal hypertension, born at term, 2 vessel cord. |
| Early motor development | Sat at 8-9 months, cruised at 12 months, walked unaided at 21 months | Crawled at 12 months, walked unaided at 24 months, clumsy from a young age | Normal (sat at 6 months, walked unaided at 9 months) |
| Early cognitive development | Normal | Lower centile in school since 4 years old | Normal |
| Unsupported walking | 21 months to present (12 years); occasional falls. | 2 years to present (11 years); clumsy, frequent falls | 9 months to present (14 years); occasional falls. |
| Presentation | |||
| Age at presentation | 3 year | 4 year 3 months | 7 year |
| Signs at presentation | Brisk reflexes, positive Babinksi bilaterally, sustained ankle clonus, short wide neck, epicanthus inversus, café au lait patches and capillary naevi | Brisk reflexes, positive Babinski bilaterally | Normal reflexes, positive Babinski bilaterally, struggles to hop and stand on one leg for long. |
| Course over time | |||
| Regression | No | Worsening gross and fine motor skills post-operative cranio-cervical decompression and meningitis | No |
| Cognition | Mild learning difficulties | Learning difficulties (1-2 years behind class, memory problems) | Normal |
| Highest motor milestone | Independent walking | Independent walking | Independent walking |
| Epilepsy | None | None | None |
| Other | Slight in-toeing gait bilaterally, mild lumbar lordosis. | Chiari 1 with craniocervical stenosis, craniocervical decompression (8 years old), post meningitic hydrocephalus with right sided VP shunt (8 years old), slight in-toeing gait bilaterally with mild lumbar lordosis. | No in-toeing, mild lumbar lordosis. |
| Physical examination | |||
| Age at latest examination | 12 years | 11 years | 14 years |
| Head circumference | Between -2 and -1 SD | Normal | Normal |
| Height | Normal | Normal | Normal |
| Weight | Normal | >2 SD | Normal |
| Vision | Myopia | Myopia | Myopia |
| Cranial nerves | Normal | Normal | Normal |
| Axial tone | Normal | Normal | Normal |
| Arms | |||
| Spasticity | No | No | No |
| Reflexes | Normal | Normal | Brisk |
| Ataxia | Yes, dysmetria | Yes, dysmetria and dysdiado-chokinesia | Yes, dysmetria and dysdiado-chokinesia |
| Extrapyramidal signs | No | No | No |
| Legs | |||
| Spasticity | Yes | None | None |
| Reflexes | Brisk | Brisk | Brisk |
| Plantar reflex | Extensor | Extensor | Flexor |
| Extrapyramidal signs | No | No | No |
| Ataxia | Unsteady on tandem gait | Unsteady on tandem gait | Unsteady on tandem gait |
| Gait | In-toeing bilaterally, mild | In-toeing bilaterally, very mild | In-toeing bilaterally, very mild |
| MRI | |||
| Age at MRI | 3 years | 5 years | 7 years |
| Signal of supratentorial white matter | |||
| Abnormal | Yes | Yes | Yes |
| Consistent with hypomyelination | Yes | Yes | Yes |
| Subcortical T1 hyperintense rim | No | No | No |
| Abnormal signal of internal capsule | No | No | No |
| Corpus callosum | |||
| Hyperintensity/thinning | No | No | No |
| Supratentorial atrophy | |||
| Generalised | No | No | No |
| Brainstem | |||
| Abnormal signal | No | No | No |
| Cerebellum | |||
| Abnormal signal / atrophy | No | No | No |
| Spinal cord | |||
| Abnormal signal of dorsal columns | Yes from C1 to C6 | Yes from C2 to C7 | Yes C1 to C6 |
| Abnormal signal of lateral corticospinal tracts | Yes from C1 to C6 | Yes from C2 to C7 | No |
| Proton MRS | |||
| Lactate elevation | Normal. MRS demonstrates possible mild elevation of choline and NAA compared to creatine. | Normal. NAA:Cr ratio 1.59 | Normal. But reduction in the normal NAA:Cr ratio 1.28. This is lower than one would expect for a child this age. |
| Other | Complex vertebral body formation abnormalities in the lumbar spine with several hemivertebrae and butterfly vertebrae causing a mild scoliosis. Trigonocephaly. | Craniocervical decompression for Chiari 1 malformation after 7 years old (2013). Mild trigonocephaly. | Very mild trigonocephaly. |
*f = female, m = male, VP = ventriculo-peritoneal shunt, SD = standard deviation, NAA = N-acetyl aspartate, Cr = creatine.
In this family, there is a variation in symptom severity and MRI findings suggesting other factors are likely involved in modifying the phenotype. Wolf et al. [9] has observed that mild viral illnesses, vaccination or injuries can cause deterioration in patients with HBSL. It is difficult to determine if Sibling 1’s deterioration in skills and behavior are solely due to meningitis or if this was compounded by having DARS mutations.
Sibling 1 had the most significant learning difficulties. Interestingly, his concentration was felt to be poor even at age 4 years. This would support Fröhlich et al. [11], that Dars+/- mice have impaired attentional processing. Sibling 1 appears to be the most affected clinically and radiologically, even prior to his meningitis.
Trigonocephaly is a feature in this sibling trio, not previously described in DARS mutations. Our proband had vertebral body abnormalities and dysmorphisms. These may be attributable to another cause as they are not found in her siblings.
The radiological features present even in the mildest forms of DARS mutations include supratentorial white matter signal abnormalities without atrophy, abnormal signal in either the brainstem, cerebellar peduncles, or spinal cord. These cases inform us of the expanding spectrum of DARS-associated HBSL and directs future research into the relationship of phenotype at a molecular and enzymatic level.
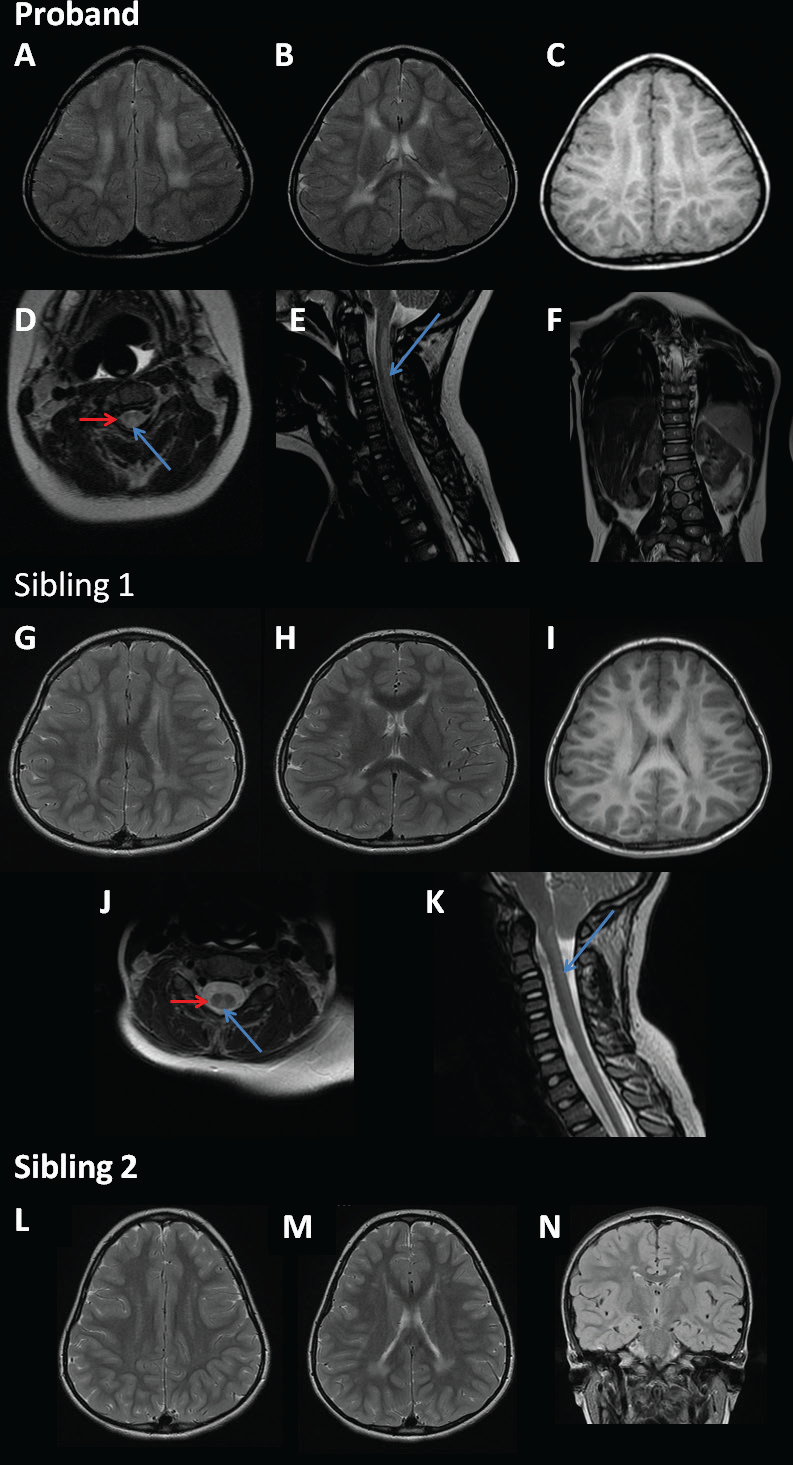
MRI pattern of siblings. MRI images of proband (images A–F), sibling 1(images G–K), and sibling 2 (images L–N). Images (A), (B), (D), (G), (H), (J), (L), and (M) are axial T2-weighted images, (C) and (I) are axial T1-weighted images, (E) and (K) are sagittal T2-weighted images, (F) is a coronal T2-weighted image, and (N) is a coronal T2-weighted FLAIR image. The axial T2 images for all three cases show high T2 signal in the supratentorial white matter and also trigonocephaly whilst (C) and (I) show intermediate and low T1 signal in the supratentorial white matter. The dorsal columns (blue long arrows) and lateral corticospinal tracts (red short arrows) of the proband and sibling 1 are hyperintense. The proband has complex vertebral body abnormalities in the lumbar spine consisting of several hemivertebrae and butterfly vertebrae.