
Background
Glutaric aciduria type 1 (GA1) is a rare autosomal recessive neurometabolic disorder characterized by abnormal accumulation of glutaric acid, 3- hydroxyglutaric acid, glutaconic acid and glutarylcarnitine in body fluids; namely blood, urine and cerebrospinal fluid (CSF). The biochemical abnormalities are secondary to deficiency of the enzyme glutaryl-CoA dehydrogenase which is essential for the catabolic pathway of the amino acids tryptophan, lysine and hydroxylysine in the mitochondria. This is caused by alterations in the glutaryl CoA dehydrogenase gene (GCDH) located on chromosome 19p13.2 [1].
Commonly, patients with this disorder have macrocephaly, developmental delay, early hypotonia and extrapyramidal features such as dystonia and other dyskinetic movement disorders. The clinical course of untreated cases is often complicated by acute encephalopathic crises precipitated by infections, fever or immunization. Subsequent neurodegeneration ensues. Early diagnosis and treatment reduce the risk of irreversible striatal degeneration and the associated movement disorder. New-born screening programs allow early identification of GA1 patients and initiation of lysine-restricted diet, carnitine supplementation with aggressive management of catabolic states with glucose infusions and low dose insulin [2].
We report a consanguineous Sudanese family of an Egyptian decent in which three members (the father, his son and daughter) were diagnosed with GA1 secondary to a novel variant sequence in the GCDH gene.
Case presentation
Family background
Parents were second-degree cousins. The father who was 33 years old, was investigated during his childhood because of gross motor and speech delay and a ‘big head’. However, no diagnosis was made at the time. He had learning difficulties, dysarthria and a degree of dyskinesia. The mother, who was in her late twenties, had no significant past medical history. She was asymptomatic. The grandparents from both sides and other family members were said to be healthy.
Index case
The index case was a 4 year old boy. He was born at term by an induced vaginal delivery following an uncomplicated pregnancy. Apart from transient jaundice, the neonatal period passed uneventfully. During early infancy, he was noted to be generally floppy with poor head support and he had minimal vocalization. At the age of 10 months, he acutely presented with irritability following a mild trauma to the head. An urgent brain CT scan showed bilateral subdural hematoma which was evacuated surgically. A few weeks later, he developed dystonia which worsened every time he had a febrile illness. Physical examination demonstrated an alert child with no dysmorphism. He had global developmental delay. Motor system examination revealed generalized dystonia with recurrent opisthotonic posturing. The occipitofrontal circumference was well above the 97th centile and his weight and length were below the 3rd centile. Other systems examination was unremarkable.
The sibling
His younger sister, who was 2 years old, was born at term by an elective Caesarean section after a normal pregnancy. She had normal motor development with good head support, normal muscle tone and normal head growth till 5.5 months of age. A couple of days after she bumped her head, she became irritable and drowsy. Her examination showed an encephalopathic infant with hypotonia and a head lag. Her anterior fontanel was tense. A brain CT scan showed minimal subdural collection with no pressure effect and she was managed conservatively. Two months later, she developed generalized dystonia and choreo-athetotic movements.
Investigations
Brain MRI scans for the two children and the father revealed bilateral fronto-temporal atrophy, dilated Sylvian fissures with open opercula (figure 1). Serum amino acids and acylcarnitine profile for both children showed increased glutarylecarnitine level and decreased free carnitine. Large amounts of glutaric acid and moderate amount of 3-hydroxyglutaric acid were detected in their urine.
Peripheral blood specimens were collected from the two siblings and their parents and genomic DNA was extracted. Sanger sequencing of the GCDH gene was performed. A pathogenic sequence variant was detected in the whole family. There was a guanine insertion in position c.1173_1174 (c.1173dupG), resulting in a frame shift and a stop codon downstream (p.Asn392Glufs4X). This duplication of guanine occurred in a DNA region of 7 guanine sequences (figure 2).
The two children and their father were found to be homozygous for this alteration. In contrast, the mother was an asymptomatic heterozygous carrier. This specific variant has not been registered in the Human Gene Mutation Database.
(https://portal.biobaseinternational.com/hgmd/pro/gene.php?gene=GCDH).
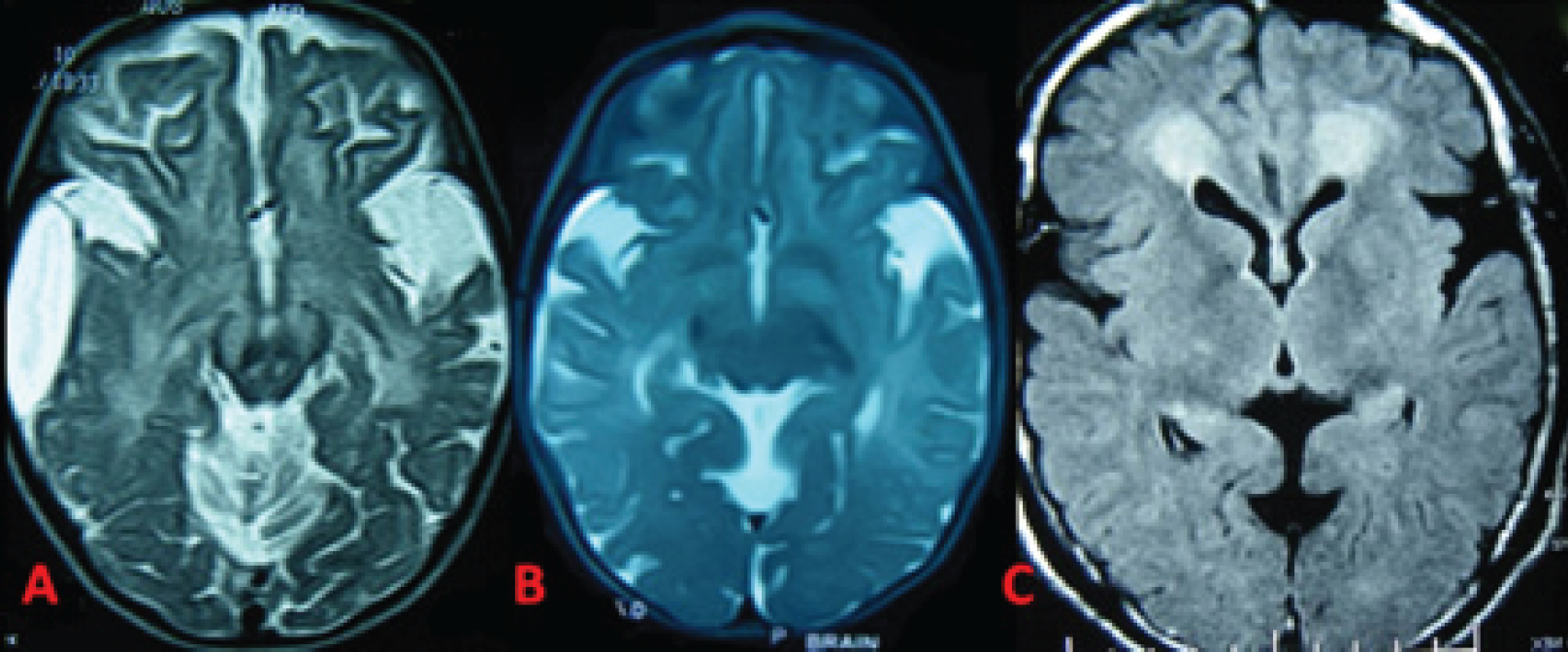
Brain MRI scans of the boy (A), the girl (B) and their father (C). There is marked atrophy with dilated Sylvain fissures more evident in the siblings compared to their father. A right subdural collection is seen in A. High signal intensity is also seen in the basal ganglia (B). Bilateral periventricular and frontal white matter T1 hyperintense lesions are more obvious in the father’s image (C).
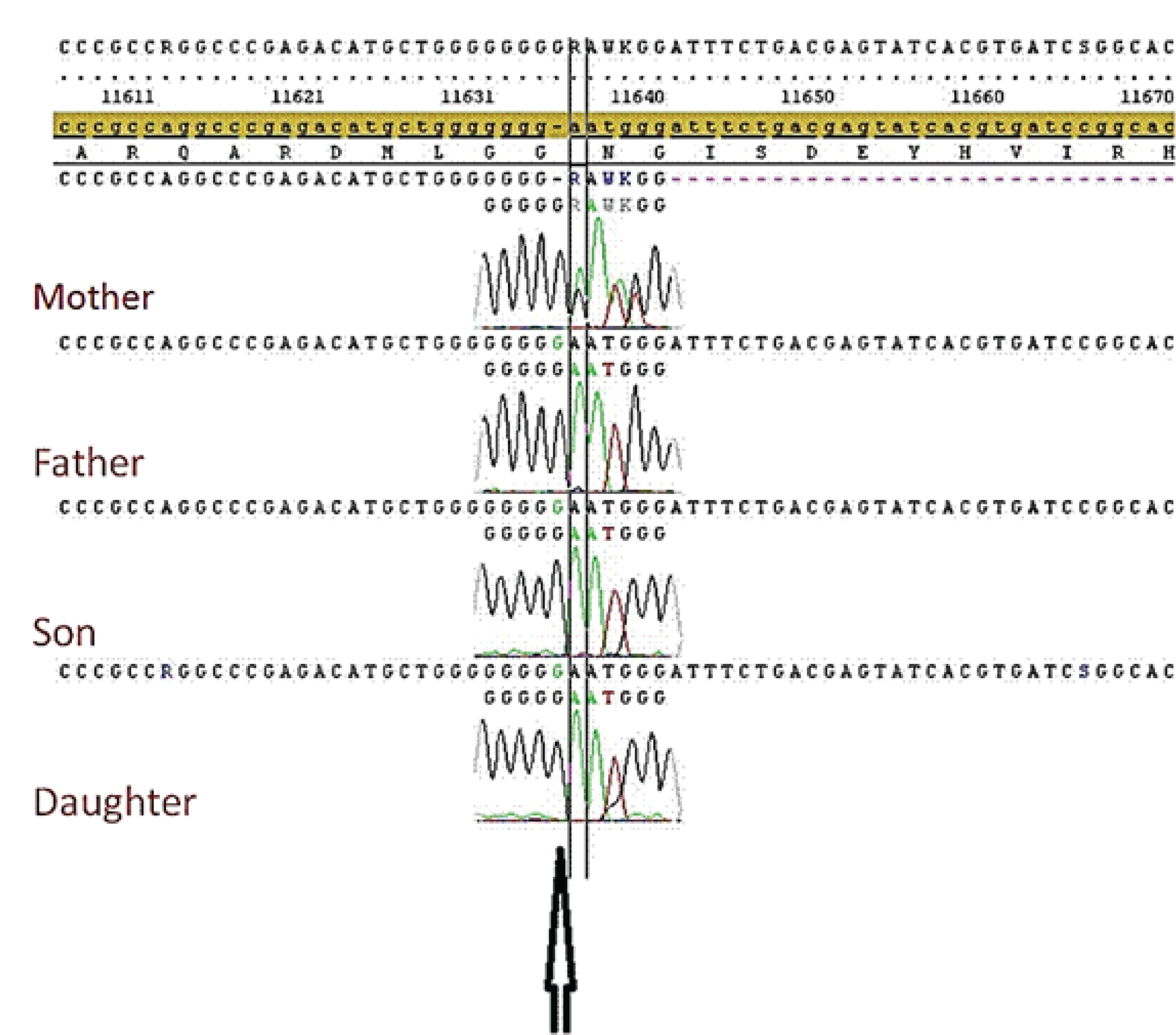
Sanger sequencing result of the GCDH gene demonstrating a guanine insertion in position c.1173_1174 as shown by the arrow. Because of this duplicated nucleotide, the following codon turns into a GAA trinucleotide rather than an AAT trinucleotide (Glutamic acid (Glu) has replaced Asparagine (Asn)) forming a change in the amino acid sequence and resulting in a frame shift and a stop codon downstream (p.Asn392Glufs4X). The top result is from the mother who is heterozygous for the mutation showing double waves; the black wave is the normal nucleotide and the green is the mutated inserted nucleotide. The bottom three results are from father, son and daughter who are all homozygous for the mutation.
Discussion
In Western countries, the estimated prevalence of glutaric aciduria type I (GA1) is 1 in 100,000 newborns [3]. It is also probable that it is second common organic acidemia in India after methylmalonic acidemia [4]. Little is known about the prevalence of GA1 in the Middle East countries where no established new-born screening programs exist. However, it can be speculated that GA1 is more prevalent in these countries due to the high frequency of consanguineous marriage and possible founder effect.
The primary defect in GA1 is the absolute or relative deficiency of glutaryl-CoA dehydrogenase which has high activity in neuronal, hepatic and renal tissues. This enzyme is coded by the GCDH gene. More than 200 different variants in the GCDH gene have been described [5]. Certain mutations are noted to be more prevalent in specific ethnicities. Most reported GA1 cases in Caucasians are compound heterozygous for two different variants. To date, the commonly reported variant in these populations, p.Arg402Try, accounts for 10% to 20% of alleles [5]. On the other hand, other variants are frequently or even exclusively seen in specific countries. Considerable variation in severity of the clinical presentation of GA1 has been observed with no correlation with the patients` genotypes.
To our knowledge, this is the first report of GA1 caused by c.1173dupG variant in a Sudanese family. Because of consanguinity in addition to the autosomal recessive inheritance of GA1, it is not surprising that the affected individuals in this family were homozygous for this variant whilst the asymptomatic mother was a carrier.
It is interesting that minor head trauma leading to subdural hemorrhages was the reason for the initial presentation of the two siblings in this family. Only a few cases of GA1 have been reported in the literature with this complication necessitating neurosurgical intervention [6].
Conclusion
Since there are no other published reports of GCDH gene variants causing GA1 in Sudanese patients, we recommend that gene testing should be undertaken in suspected cases. This will help improve identifying the common variants in these communities. Moreover, the institution of new-born screening programs in these countries will help improve the outcome for these patients. Genetic testing should be made widely available in communities where consanguinity is prevalent. This will allow detection of new pathogenic gene variants.