
Background
Herpes simplex virus 1 and 2 and varicella zoster virus are closely related, double-stranded DNA viruses which belong to the alphaherpesvirus subfamily of Herpesviridae [1]. They are composed of 162 capsomeres arranged in an icosahedral structure and can be distinguished via immunofluorescence, immunohistochemistry, in situ hybridization and PCR [2]. HSV and VZV share several features including sensory neurotropism, the ability to establish durable latency in the trigeminal and dorsal root ganglia, and the ability to reactivate resulting in episodic outbreaks. They cause similar but clinically distinct vesicular mucocutaneous eruptions reflective of their disparate mechanisms of pathogenesis and latency [3,4]. During initial infection, virions enter the sensory neurons in the epithelium and travel via retrograde axonal transport to the associated sensory ganglion [2]. Given the wide anatomic distribution of primary infection in varicella, VZV DNA can be distributed hematogenously to any sensory ganglion as well as to autonomic (i.e. enteric) ganglia that do not innervate the periphery [3]. Reactivation triggers anterograde movement of the virus and either asymptomatic shedding or recurrent disease [5].
The rates, timing and precipitating factors for reactivation of HSV and VZV differ considerably [1]. It has been estimated that HSV-1 seropositivity approaches 90% by age 60 [4]. HSV-2 seropositivity exceeds 16% overall, with recurrent genitourinary lesions seen in approximately 20 to 50% and minor or atypical lesions seen in half of other seropositive individuals. Episodic asymptomatic shedding is possible at a low rate even in those without clinically apparent recurrences. HSV occurs more often in the young and may reactivate in more than half of seropositive individuals up to hundreds of times in a lifetime [1]. Incidence of recurrence declines throughout life, consistent with an evolving immune response. Patients with frequent episodes of herpes labialis tend to have higher levels of HSV-specific antibodies but lower levels of interferon gamma and interleukin 2, suggesting that cytokine release may play greater role than immunoglobulins in suppressing herpesvirus reactivation [3].
While VZV seropositivity is also greater than 90%, estimated lifetime risk of zoster is only 10-20%. VZV usually reactivates only once and rarely more than once. The incidence of zoster increases with advancing age, corresponding to a decline in the VZV-specific T lymphocyte population [4]. Humoral immunity appears to have minimal effect on the rate and severity of reactivation as zoster occurs despite adequate antibody levels and occurs with normal frequency in agammaglobulinemic individuals.
As HSV and VZV both establish latency in sensory nerve ganglia, autopsy studies have predictably demonstrated the presence of both viruses in the same ganglia, even within the same neurons [6]. But despite their potential co-localization within the ganglion, the two viruses rarely cause simultaneous disease. In the majority of these cases, the viruses have been isolated at different body sites [7,8] and in the setting of immunosuppression [7,9,10]. This is consistent with the observations that immunosuppressive conditions or treatments which impair cellular immunity can cause more frequent and severe reactivation by either virus [4]. Simultaneous dissemination of one or more herpesviruses with or without localized disease has also been reported [7,9-11]. Clinical disease with concurrent detection of both viruses from the same anatomic location, even in immunocompetent hosts, is more rarely described in the literature [1,2,5,12]. Here we report a case of VZV and HSV-2 co-reactivation involving the penis in an otherwise immunocompetent patient, with poorly controlled diabetes mellitus as the apparent precipitating factor. To our knowledge this is the first case of diabetes-related concurrent herpesvirus reactivation.
Case Presentation
A 64-year-old male presented with a one-week history of penile pain, swelling and discharge. Swelling preceded gradually worsening pain and serous discharge by approximately three days. Pain was partially relieved with the application of petroleum jelly. He denied any history of sexually transmitted infections and prior episodes of penile discharge or genital ulcers. He was sexually active with his wife only. Review of systems was positive for subjective fever and chills during the past week as well as baseline exertional dyspnea and distal paresthesias. Past medical history was significant for type 2 diabetes mellitus (DM), hypertension (HTN), chronic kidney disease (CKD) stage III, mild systolic congestive heart failure (CHF), chronic obstructive pulmonary disease (COPD), and stage IIIb lung cancer for which he completed treatment with chemotherapy (carboplatin and pemetrexed) and radiation approximately nine months prior to presentation. A recent computed tomography (CT) scan of the thorax was negative for recurrent disease. Family history included a brother with lung cancer.
At presentation, the patient was afebrile with a blood pressure of 142/82 mmHg, heart rate of 93 beats/minute, and SpO2 of 95% on room air. He was alert, fully oriented, and appeared to be in no acute distress. Physical exam revealed an uncircumcised phallus with multiple small, shallow, clean based ulcers on the foreskin. Retraction of the foreskin was not possible due to the moderate degree of pain elicited. Minimal odorless, serous drainage from the ulcers was noted. Four subcentimeter, crusted lesions were also observed in a linear pattern at the left inferolateral abdomen in the L1 dermatome. The remainder of the exam was unremarkable.
In the emergency department (ED), the patient was found to be markedly hyperglycemic at 577 mg/dL (74-106 mg/dL). There was associated pseudohyponatremia with a serum sodium of 125 mmol/L (136-145 mmol/L) however bicarbonate, lactic acid and anion gap were within normal limits. Creatinine was found to be 2.1 mg/dL (0.3-1.2 mg/dL), which was near the patient’s baseline. Hemoglobin A1c was increased to 14.9% (4.16-4%) from 8.7% two months earlier. His white blood cell count, platelets, and hematocrit were all within normal limits. Urinalysis showed pyuria (10-20 leukocytes/HPF [0-5]) and glycosuria (>1000 mg/dL [0 mg/dL]) but was negative for bacteria. Urine culture, urine Gonorrhea/ Chlamydia nucleic acid amplification tests (NAATs), rapid plasma reagin (RPR), and fourth generation HIV test were negative.
The patient’s blood glucose and sodium improved with subcutaneous insulin and IV fluids. He was admitted to the general medical floor for further workup. Empiric ceftriaxone and metronidazole which had been started in the ED were stopped. Oral acyclovir was started at a dose of 400 mg TID for suspected genital herpes. Swabs were collected from the base of a penile ulcer and sent for both polymerase chain reaction (PCR) and viral culture. The sample was positive for both VZV and HSV-2 DNA by PCR. The dose of acyclovir was increased to 800 mg five times daily as appropriate for the treatment of zoster. The cycle threshold (Ct) for VZV was 35.3 while the corresponding value for HSV-2 was 40. The viral culture later grew HSV-2 as well. Serologies indicated prior exposure to HSV-1, HSV-2 and VZV.
On discharge, acyclovir was changed to oral valaciclovir at a dose of 1 gram three times daily (TID), the higher dose indicated for zoster. The patient’s genital ulcers had resolved on follow-up with his primary care physician. His glycemic control also improved with increased insulin dose and closer adherence to a diabetic diet. Repeat hemoglobin A1c at three months had decreased to 8.8%.
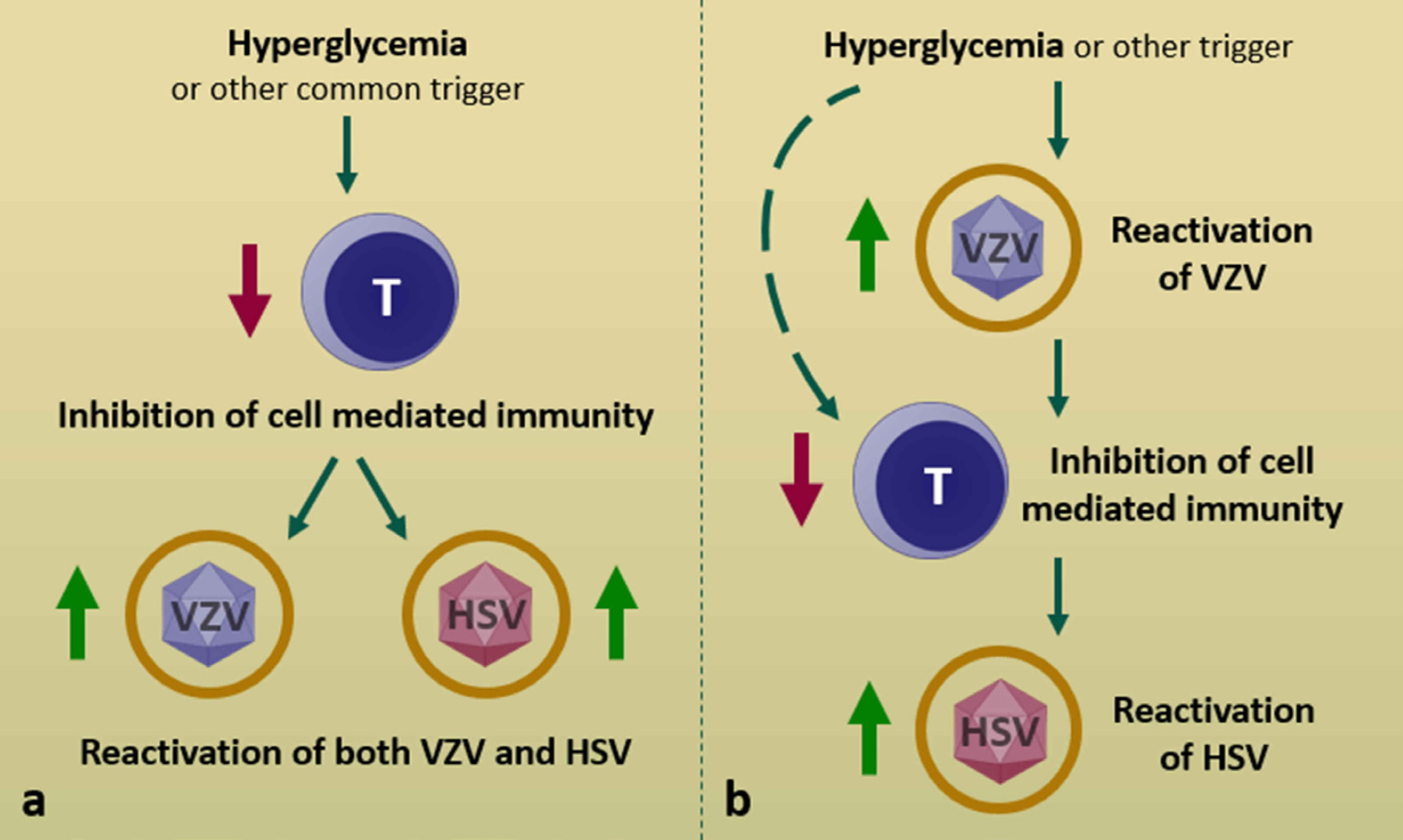
Models for concurrent herpesvirus reactivation. (a) A common triggering event (e.g. hyperglycemia) reactivates two viruses within the same ganglion at once. (b) Reactivation of one virus leads to further inhibited cell mediated immunity and subsequent reactivation of another latent virus. It is also plausible that hyperglycemia-induced T cell inhibition allows both HSV and VZV to escape immune control, but VZV has a propensity to reactivate first (dashed line).
Discussion
The location of herpesvirus latency within the ganglion is of paramount importance with regard to their reactivation and clinical manifestations. HSV localizes exclusively to the neuron [6]. This unique characteristic explains its ability to reactivate frequently in response to a variety of triggers including physical and emotional stress, ultraviolet light, trauma or physiologic factors such as changes in sex hormones levels [1]. During latency, most HSV gene expression is silenced due to maintenance of DNA in a heterochromatin state [3]. However, latency associated transcripts (LATs) are abundantly produced which increase the efficiency of latency establishment and reactivation via their ability to inhibit both neuronal apoptosis and lytic gene expression. Interestingly, LATs also interfere with superinfection by other strains of the same virus or by other herpesviruses [6]. Therefore, dual infection of the same neuron with HSV and VZV appears to be a rare, stochastic event.
There is minimal, if any, interneuronal spread of HSV prior to anterograde axonal transport to the periphery, reflective of its characteristic localized mucocutaneous lesions [3]. Zosteriform presentations of HSV have however been described suggesting that interneuronal spread is possible [1]. In contrast, VZV demonstrates extensive intraganglionic spread due to its residence within satellite cells. Zoster-associated neuralgia is likely a consequence of the extensive cytopathology associated with this process [4]. The large, dermatomal lesions seen in herpes zoster reflect peripheral delivery by multiple neurons surrounded by the involved neuroglia [3,8].
VZV is less easily reactivated than HSV due to the requirement for intercellular signaling between the satellite cell and the neuron [1]. Trauma and x-ray have been implicated as triggers anecdotally, however VZV has proven difficult to experimentally reactivate from human ganglia [3,4]. The mechanisms of latency and reactivation of VZV are less well understood than those of HSV due to a relative lack of in vitro or animal models. It appears that VZV lacks the equivalent of LATs but expresses some lytic transcripts during latency, namely ORF63 [3]. The protein encoded by ORF63 modulates interferon type I activity and inhibits apoptosis of the infected neuron. HSV-specific CD8+ cells have been shown to infiltrate murine sensory ganglia during acute infection and remain apposed during latency, supporting the concept that viral reactivation is mediated by transient inhibition of T cell function [3]. VZV specific T cell infiltrates have not been identified histologically but may play a similar role given the clear importance of cellular immunity in VZV reactivation.
Two models for concurrent herpesvirus reactivation have been posited. Either primary infection with or reactivation of one virus leads to decreased cell mediated immunity and subsequent reactivation of another latent virus, or a common triggering event reactivates two viruses within the same ganglion at once (Figure 1) [1,8]. Two retrospective studies have examined concurrent detection of HSV and VZV from the same anatomic site. Giehl et al. found that, of 1718 patients with lesions suspicious for herpesvirus infection, 20 (1.16%) were coinfected at the same site [2]. The face was most commonly involved and there were no significant differences with regard to age or sex. This study did not differentiate between HSV-1 and HSV-2 however the genital region was only involved in one patient. Most patients reported personal stress prior to the onset of disease and two thirds had a history of recurrent labial or genital infections. Another 8-year retrospective study also surveyed concurrent HSV and VZV at the same site [5]. Of 8249 samples positive for one or both viruses, 108 (1.3%) were dually positive. Unlike the earlier study, this one distinguished between HSV subtypes; two thirds of samples were positive for VZV and HSV-1 whereas just over a quarter were positive for VZV and HSV-2. The most commonly involved sites were the face, lips, oral mucosa, and genitourinary regions, consistent with the ratio of HSV subtypes isolated. From the limited data available, there appears to be no concern for viral antagonism. Also, the courses of herpes simplex and zoster were are not altered when occurring simultaneously [2].
The work of Dhiman et al., in particular, shed light on the pathogenesis of concurrent herpesvirus reactivation [5]. They consistently observed a lower cycle threshold (Ct) for VZV compared with HSV, indicating a higher burden of VZV at the site of infection. This suggests that the development of zoster was the primary event that caused secondary reactivation of HSV. Our patient also had a lower Ct for VZV compared with that for HSV-2. Furthermore, since he had a separate resolving zoster rash of the abdomen, it correlates clinically that he experienced reactivation of VZV in the L1 dermatome prior to reactivation of both HSV-2 and VZV in his genital area (S3 dermatome).
The precipitating factor for the development of zoster in our patient appears to have been poorly controlled diabetes mellitus. Any immunosuppressive effect of the chemotherapy he received for lung cancer would no longer be expected to contribute as the course ended approximately nine months prior to presentation. Inhaled corticosteroids used for COPD do not achieve clinically relevant systemic concentrations. The patient had not experienced any other known stressors or traumas. Hyperglycemia is known to cause reversible T lymphocyte dysfunction [13]. Given that waning cellular immunity has been implicated in zoster, it is unsurprising that diabetes has been associated with increased incidence of zoster and post-herpetic neuralgia [14]. At the same time, there is merit to the hypothesis that a single common trigger may be responsible for reactivation of both HSV and VZV in coinfected patients. Some studies have identified HSV seropositivity as a risk factor for diabetes, possibly due to chronic inflammation or immune activation [15]. It is plausible that hyperglycemia-induced T cell inhibition allows both HSV and VZV to escape immune control, but that VZV simply has a propensity to reactivate first.
Conclusion
Concurrent reactivation of HSV and VZV at the same anatomic site is an uncommon but well-described event. The clinical significance of codetection is unknown. The development of zoster appears to facilitate the subsequent reactivation of HSV based on Ct data, though common triggers that globally suppress T cell function may play a role in reactivation of both viruses within the same ganglion. If clinical examination raises a suspicion for both VZV and HSV, it is prudent to test for both because higher doses of antiviral agents are recommended for the successful treatment of herpes zoster as compared to herpes simplex infections. In our patient, valacyclovir at a higher dose (1 gram TID) proved to be an effective outpatient oral treatment option. Clinicians should be cognizant of the possibility of simultaneous HSV and VZV infection in patients presenting with zosteriform lesions, particularly those with uncontrolled diabetes or who are immunocompromised.