
NON-STANDARD ABBREVIATIONS AND ACRONYMS
ACEI | angiotensin-converting enzyme |
CPMR | chronic primary mitral regurgitation |
ECM | extra-cellular matrix |
EMB | endomyocardial biopsy |
IQR | interquartile range |
LV | left ventricular |
MMP | matrix metalloproteinase |
NT-pro-BNP | N-terminal-pro hormone B-type natriuretic peptide |
RNA | ribonucleic acid |
RV | right ventricular |
ST2 | suppression of tumourigenicity 2 |
TAPSE | tricuspid annular plane systolic excursion |
TIMP | tissue inhibitor of matrix metalloproteinases |
TNF | tumour necrosis factor |
INTRODUCTION
Chronic primary mitral regurgitation (CPMR) is caused by the progressive degeneration of the mitral valve apparatus in developed countries and by rheumatic heart disease in developing countries. (1,2) Patients with CPMR are often asymptomatic for many years while the left ventricle (LV) progressively remodels in response to abnormal preload. The eccentric hypertrophy that develops normalizes afterload but is insufficient to fully compensate for the wall stress.(3) This leads to an increase in inflammatory cytokine expression,(4–7) neuro-hormonal activation (8) and reactive oxygen species generation.(9) These chronic stressors activate a number of complex inflammatory and apoptotic pathways in a similar manner to heart failure from other causes.(10) Ultimately, there is myocyte loss and sliding displacement of cardiomyocytes, or cell slippage, caused by disruption of the myocardial extracellular matrix (ECM)-integrin linkages.(11,12) Later changes include myocyte apoptosis and pathological ECM fibrosis, leading to cardiac decompensation.(13) At present, there are no commercially available markers of adverse left ventricular (LV) remodelling, which could be used to guide the optimal timing of surgical intervention.(14) By the time a decompensated stage is reached in the progression from compensated to decompensated CPMR, there may be irreversible pathological fibrosis and myocyte loss that cannot be corrected by surgical intervention.
We recently described important differences in the gene expression signature of adverse LV remodelling in patients with compensated and decompensated CPMR,(15) supporting previous work in a rat model of volume overload.(16) Early compensated CPMR appears to be characterized by preserved LV ejection fraction and an increase in beta-adrenergic drive.(17–19) Myocardial expression of genes important in ECM and inflammation are normal during early compensated CPMR, while persistent volume overload increases the expression of the genes coding for atrial natriuretic peptide and NT-pro-BNP (N-terminal-pro hormone B-type natriuretic peptide). Decompensation is characterized by LV systolic dysfunction, an elevated serum NT-pro-BNP and abnormal expression of genes important in calcium regulation, inflammation, apoptosis, fibrosis and mitochondrial function.(5,15,16,20)
Although several molecular markers of decompensated CPMR have been identified,(15,16,19,21) whether the circulating proteins of these markers can be used as biomarkers of decompensation depends on their correlation with the gene expression levels and the clinical phenotype. We previously showed that serum NT-pro-BNP correlated with clinical markers of decompensated CPMR and with higher serum levels of tissue inhibitor of matrix metalloproteinases (TIMP)-1, matrix metalloproteinase (MMP)-2, soluble FAS and suppression of tumourigenicity 2 (ST2) and inversely correlated with serum MMP-9.(15) Here we compare and correlate several serum markers of adverse LV remodelling with the myocardial expression levels of their respective genes.
METHODS
Study subjects
The investigation conforms to the principles outlined in the Declaration of Helsinki. Informed consent was obtained from all patients according to a protocol approved by the Human Research Ethics Committee of the University of the Witwatersrand, Johannesburg, South Africa (M101036). Patients were included in the study if they had CPMR and met guideline criteria for elective mitral valve surgery.(14) Patients were excluded if they had aortic valve disease, mitral stenosis, active infective endocarditis, co-morbid coronary artery disease, systemic hypertension, chronic kidney disease, contraindications to β-blocker therapy, were pregnant or had a history of anti-remodelling therapy use (β-blockers, angiotensin converting enzyme inhibitors, angiotensin receptor blockers or mineralocorticoid receptor antagonists).
Twelve patients underwent baseline clinical examination, transthoracic echocardiography, blood analysis and cardiac catheterization, during which LV endomyocardial biopsies (EMBs) were obtained using disposable 2.2 mm × 105 cm Jawz® Endomyocardial Biopsy Forceps (Argon Medical, TX, USA) as previously described.(15) Biopsy specimens were frozen in liquid nitrogen for gene expression analysis and collected in 10% buffered formalin for histopathology. The frozen specimens were then stored at -80°.
Echocardiography
Transthoracic echocardiography was performed with an iE-33® ultrasound device (Koninklijke Philips, NV, USA) by a single sonographer on all recruited patients according to established guidelines.(22)
Serum
Quantification was performed using Magnetic Luminex® Screening Assays (R&D Systems, Inc., MN, USA) [for tumour necrosis factor (TNF)α, Interleukin (IL)-1β, IL-6, ST2, FAS-ligand and FAS] or Milliplex® MAP panels (EMD Millipore Corporation, MA, USA) (for MMP-1, -2 and -9, and TIMP-1 and -4). Assays were run using the Bio-Plex™ 200 system (Bio-Rad, TX, USA) and concentrations generated automatically using the Bio-Plex manager software, version 5.0 (Bio-Rad Laboratories Inc., Hercules, CA, USA).
RNA isolation
Ribonucleic acid (RNA) was extracted using the miRNeasy Mini Kit® (Qiagen Sciences, MD, USA), quantified using spectrophotometric analysis on a NanoDrop ND-2000 Spectrophotometer (NanoDrop Technologies Inc., Wilmington, Del.), and RNA integrity was determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA).
Gene expression and statistical analysis
Multiplex gene expression analysis, using 100 ng total RNA per sample, was performed commercially by NanoString Technologies (https://www.nanostring.com/scientific- content/technology-overview/ncounter-technology) using an nCounter® Custom Codeset (Seattle, WA, USA). The identity and description of the genes selected for analysis are given in Table 1. The analysis was performed on the nCounter platform using the NanoString nCounter technology.(23) Results were analysed using the NanoString® nSolver Analysis Software Package 3.0®. A background subtraction using the geometric mean of the negative controls was included, and gene expression levels were normalized to both the geometric mean of the positive controls and the geometric mean of three reference genes (GAPDH, PGK1 and HPRT1). Spearman correlation was used to measure the correlation between serum markers and the expression levels of their respective genes. A P-value <0.05 was considered to be statistically significant.
Gene names, accession numbers and description of the markers
| Gene name | Accession number | Description |
| NPPB | NM002521.2 | Brain natriuretic peptide (BNP) |
| IL1R1 | NM001320984.1 | Suppression of tumorigenicity 2 (ST2) |
| FAS | NM152876.1 | FAS cell surface death receptor |
| FASLG | NM000639.1 | FAS ligand |
| TNF | NM000594.2 | Tumour necrosis factor α |
| IL-6 | NM000600.3 | Interleukin 6 |
| MMP-1 | NM002421.3 | Matrix metalloproteinase 1 |
| MMP-2 | NM004530.2 | Matrix metalloproteinase 2 |
| MMP-9 | NM004994.2 | Matrix metalloproteinase 9 |
| TIMP-1 | NM003254.2 | Tissue inhibitor of matrix metalloproteinase 1 |
| TIMP-4 | NM003256.2 | Tissue inhibitor of matrix metalloproteinase 4 |
RESULTS
Clinical, echocardiographic and haemodynamic characteristics of the 12 patients with CPMR are listed in Tables 2 and 3. Spearman correlations of the serum and gene expression levels for NT-pro-BNP, ST2, MMP-1, MMP-2, MMP-9, TIMP-1, TNFα and FAS are shown in Figure 1. Correlation coefficients for serum and gene expression levels of IL-6 and FAS ligand were −0.148 (P = 0.635) and 0.133 (P = 0.66), respectively. Figure 2 summarizes the correlation coefficients for each of the markers compared with the respective significance values. Overall, there was no significant correlation between the serum levels and gene expression levels for the markers evaluated in this study.
Baseline clinical characteristics (n = 12)
| Duration of follow-up (months) | 8.3 (±4.8) |
| Age (years) | 39 (±14) |
| Female gender | 8 |
| Black African | 11 |
| Height (cm) | 169 (±9) |
| Weight (kg) | 67 (±13) |
| Body surface area (m2) | 1.8 (±0.2) |
| Atrial fibrillation on warfarin | 2 |
| NYHA classification | 2 (2, 2) |
| Heart rate (beats per minute) | 78.8 (±16) |
| SBP (mmHg) | 116 (±13) |
| 6MWT (m) | 435 (±67) |
| NT-pro-BNP (ng/L) | 619 (107, 1824) |
NYHA = New York Heart Association, SBP = systolic blood pressure, 6MWT = 6-min walk test. Values are means ± standard deviations except for NYHA and NT-pro-BNP, where medians and interquartile ranges are reported.
Baseline echocardiographic and haemodynamic characteristics
| Echocardiography | |
| LVEDD (mm) | 63 (±10) |
| LVESD (mm) | 44 (±9) |
| LVEDV indexed (mL/m2) | 123 (±42) |
| LVESV indexed (mL/m2) | 53 (±22) |
| LA volume indexed (mL/m2) | 127 (±48) |
| Ejection fraction (%) | 57 (±9) |
| LV dp/dt (mmHg/s) | 1306 (±495) |
| GLS (mm) | −16 (±5) |
| TAPSE (mm) | 18 (±4) |
| TR V max (mmHg) | 38 (±18) |
| Cardiac catheterization | |
| Right atrial mean (mmHg) | 8 (±6) |
| RV SBP (mmHg) | 53 (±25) |
| RV EDP (mmHg) | 5 (4, 8) |
| PASP (mmHg) | 53 (±23) |
| PA mean pressure (mmHg) | 37 (±16) |
| PCWP (mmHg) | 25 (±7) |
| LV EDP (mmHg) | 16 (±8) |
| LV SBP (mmHg) | 115 (±15) |
| Aortic SBP (mmHg) | 115 (±17) |
| Cardiac output (L/min) | 3.6 (±1.0) |
| Indexed CO (L/min/m2) | 2.0 (±0.5) |
LVEDD = LV end diastolic diameter, LVESD = LV end systolic diameter, LVEDV = LV end diastolic volume, LVESV = LV end systolic volume, GLS = global longitudinal strain, TAPSE = tricuspid annular plane systolic excursion, TR V max = tricuspid regurgitation maximum velocity, SBP = systolic blood pressure, RV = right ventricle, EDP = end diastolic pressure, PASP = pulmonary artery systolic pressure, PA = pulmonary artery, PCWP = pulmonary capillary wedge pressure, LV = left ventricle, CO = cardiac output. Values are means ± standard deviations except for RV EDP, where median and interquartile range are reported.
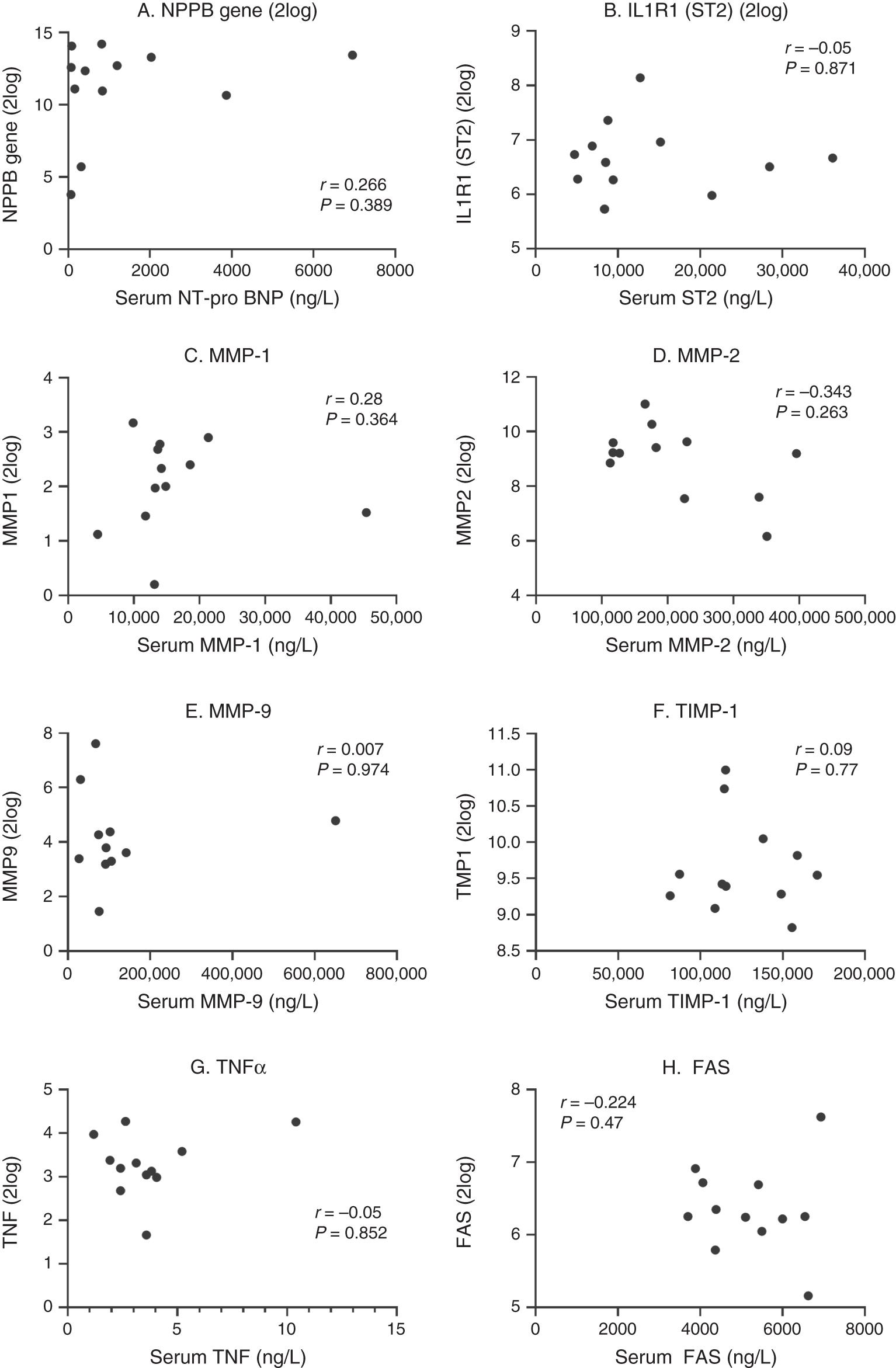
Correlations between serum and tissue gene expression levels for NT-pro BNP (A), ST2 (B), MMP-1 (C), MMP-2 (D), MMP-9 (E), TIMP-1 (F), TNFα (G) and FAS (H) for the 12 patients with CPMR. Values for gene expression levels are given in 2log change compared to reference gene levels.

Correlations were computed between serum protein concentration (ng/L) and gene expression (2log value) using the Spearman test. The X-axis presents the correlation and the Y-axis shows the P-value. A strong and significant correlation stands when the value falls within the boxes indicated with hatched marking with a P-value < 0.05 and correlation >0.8 or >−0.8.
DISCUSSION
Identification of markers of adverse cardiovascular outcomes is useful for both research and clinical application. Serum NT-pro-BNP, for example, is a marker of heart failure that is routinely used in clinical practice to aid in the diagnosis of heart failure and to risk-stratify patients presenting with heart failure.(24) Several other markers of adverse LV remodelling have been studied in patients with valvular heart disease and heart failure in including ST2, cytokines, markers of apoptosis (FAS and FAS ligand) and the MMPs and their inhibitors.(25–32) Here we have shown that there is a poor correlation between the tissue gene expression levels of several of these markers of adverse LV remodelling in patients with CPMR and the serum levels of these markers sampled at the same time.
We have previously reported that serum NT-pro-BNP correlated with clinical markers of decompensated CPMR.(15) Furthermore, serum NT-pro-BNP strongly correlated with higher serum levels of TIMP-1, MMP-2, soluble FAS and ST2 and inversely correlated with serum MMP-9. Previous studies in peripartum cardiomyopathy and dilated cardiomyopathy have demonstrated that serum FAS levels correlate with LV dysfunction.(26,33) In patients with aortic stenosis, mitral regurgitation and heart failure, circulating levels of cytokines are increased.(4,26,34) However, no previous study has simultaneously correlated the myocardial gene expression levels of these markers with the serum levels in patients with CPMR.
These data suggest that in order to fully understand serum levels of these markers, more detailed analysis at the transcriptional and post-transcriptional levels is required in patients with CPMR. MMPs, for example, are critically important in irreversible remodelling of the ECM in CPMR and are tightly regulated at various levels from gene to circulating protein.(35) MMPs and TIMPs are expressed and secreted into the extracellular space by a variety of cells, including cardiac myocytes, cardiac fibroblasts and macrophages.(36) In response to different haemodynamic overloads (pressure versus volume), the ECM undergoes different patterns of remodelling.(13) Volume overload results in loss of interstitial collagen,(21) whereas pressure overload increases fibrogenic factors and collagen deposition.(37,38)
Evidence suggests that MMP gene expression is predominantly regulated at the transcriptional level.(39) Several MMPs share promoter elements, which means that they are often co-expressed in response to inductive factors (such as cytokines).(40) For example, MMP-1 and MMP-9 share a common promoter that binds Jun and Fos transcription factors meaning that the induction of transcription of the MMP-1 would also result in transcription of MMP-9. Expressed mRNA is then translated into inactive pro-zymogens, which are secreted into the extracellular space. Unlike MMP-1, MMP-9 is stored in neutrophils,(35) which will influence the serum levels of this MMP depending on tissue inflammation. Most data on zymogen activation comes from in vitro data that may not be applicable in humans. Nevertheless, proteolytic activation of MMPs is regulated by many different agents, including thrombin and plasmin,(41) and also influenced by local factors such as hypoxia.(42) Furthermore, once activated, the MMPs can be inhibited by TIMPs, which are also regulated by numerous ECM factors.(43) Similarly, the serum levels of NT-pro-BNP depend on a number of pre- and post-translational regulatory steps.(44) Because of these complexities, it is not surprising that we did not find a correlation between gene expression levels and serum levels for these factors. A lack of correlation between myocardial tissue MMPs and serum MMPs has been described in a rabbit model of cardiomyopathy (45), and a poor correlation between peripheral blood cell RNA levels of MMP-2 and -9 and serum levels of these markers has also been described in humans with chronic active hepatitis C.(46) Finally, although there is evidence to suggest that the myocardium is an important source of MMPs, it is not possible to exclude other sources.(47,48) Activation of MMP pathways in other organs may explain the increased circulating levels of MMPs in these patients.
CONCLUSION
Here we have shown that there is a poor correlation between the myocardial gene expression levels of several markers of adverse LV remodelling in patients with CPMR and the serum levels of these markers sampled at the same time. The factors governing the expression of specific MMPs in the myocardium and whether single serum markers can be used to accurately determine the degree of adverse LV remodelling in CPMR or not needs further clarification.
