
INTRODUCTION
Monkey B virus (BV), currently designated as Macacine alphaherpesvirus 1 by the International Committee on Taxonomy of Viruses [1], is a member of the subfamily Alphaherpesvirinae [2]. Together with human herpes simplex virus types 1 and 2 (HSV-1 and HSV-2, respectively), BV constitute the genus, Simplexvirus [2]. The natural host of BV is the Asian monkey of the genus, Macaca, which generally manifests as mild, localized or asymptomatic infections [2–4]; however, BV infection in humans and other macaques species has a relatively high mortality rate [1,2]. In addition, human-to-human transmission of BV has been reported, indicating a risk of secondary transmission [5]. Because BV infects humans through bites, scratches, and other injuries inflicted by macaques [6,7], veterinarians, animal care personnels, and laboratory researchers are the main groups at risk of BV infection [8].
BV is a linear, double-stranded DNA (dsDNA) virus, the genomes of which range in size from 154,958–157,447 bp [1]; the coding sequences are usually conserved [9,10]. The sequence variation among BV strains of different macaque species results in various genotypes of BV [9]. The US6 gene encodes the gD glycoprotein, which is highly conserved among primate viruses and has no significant genotypic variation among BV genotypes [11]. Moreover, the UL27 gene encoding gB and gD glycoproteins (US6 gene product), are structurally conserved and have BV antigenic specificity [12–15].
Research involving diagnostic methods for BV has not been promoted. Most available diagnostic methods were developed several years ago and are not associated with advanced technologies. Scinicariello et al. [16] and Perelygina et al. [17] developed a polymerase chain reaction (PCR)- and TaqMan-based real-time PCR assay, respectively, for clinical investigation and testing, and the methods used in subsequent BV studies were usually based on PCR technology [1,14,18,19]. These diagnostic methods require sophisticated instruments and time-consuming processes for detection, so a simple, rapid, and sensitive method for the detection of BV nucleic acids is needed.
Recombinase polymerase amplification (RPA) is a novel isothermal amplification technology that does not require sophisticated equipment and is easy to operate. In the current study, we developed two RPA assays for BV nucleic acid detection. The data showed that both assays have low detection limits after a full-process detection of 30 min. In addition, a closed vertical flow (VF) visualization strip device was used for result determination, which reduces the possibility of nucleic acid contamination after amplification.
METHODS
Rationale of RPA-VF assay
The RPA combined with a VF visualization strip device principle was described in detail in a previous report [20]. Briefly, the specific probe was labeled with fluorescein isothiocyanate (FITC) and a C3-spacer at the 5′ and 3′ ends, and a tetrahydrofuran (THF) residue was used to replace a nucleotide. In addition, the reverse primer was labeled with biotin at the 5′ end. Recombinase, single-stranded binding (SSB) proteins, and strand-displacing DNA polymerase trigger primer extension events to synthesize double-stranded DNA for amplification. As a result, the amplicons were labeled by FITC and biotin. Then, a closed VF visualization strip device was used to visualize the amplification products.
RPA-VF reaction
A recombinase-aid amplification kit (Hangzhou ZC Bio-Sci&Tech Co., Ltd., Hangzhou, China) was used to demonstrate the RPA-VF reaction according to the manufacturer’s instructions. Briefly, a 47.5-μL solution (25 μL of rehydration buffer containing enzymes, 17.9 μL of diethyl pyrocarbonate [DEPC] water, 2 μL of template, 2 μL of the forward and reverse primers [each at 10 μM], and 0.6 μL of a 10 μM probe) was prepared. Then, 2.5 μL of 280 mM magnesium acetate (MgAc) was added to the reaction system. The mixture was incubated at the indicated temperatures for the specified time. Finally, the result was read using a closed VF visualization strip device (Ustar Biotech Co., Ltd., Hangzhou, China). A positive result was observed as two red bands at the test and control lines on the strip; if only one band at the control line appeared, the result was considered negative. The experimental flow of the RPA-VF assay and VF visualization strip is shown in Fig 1A.
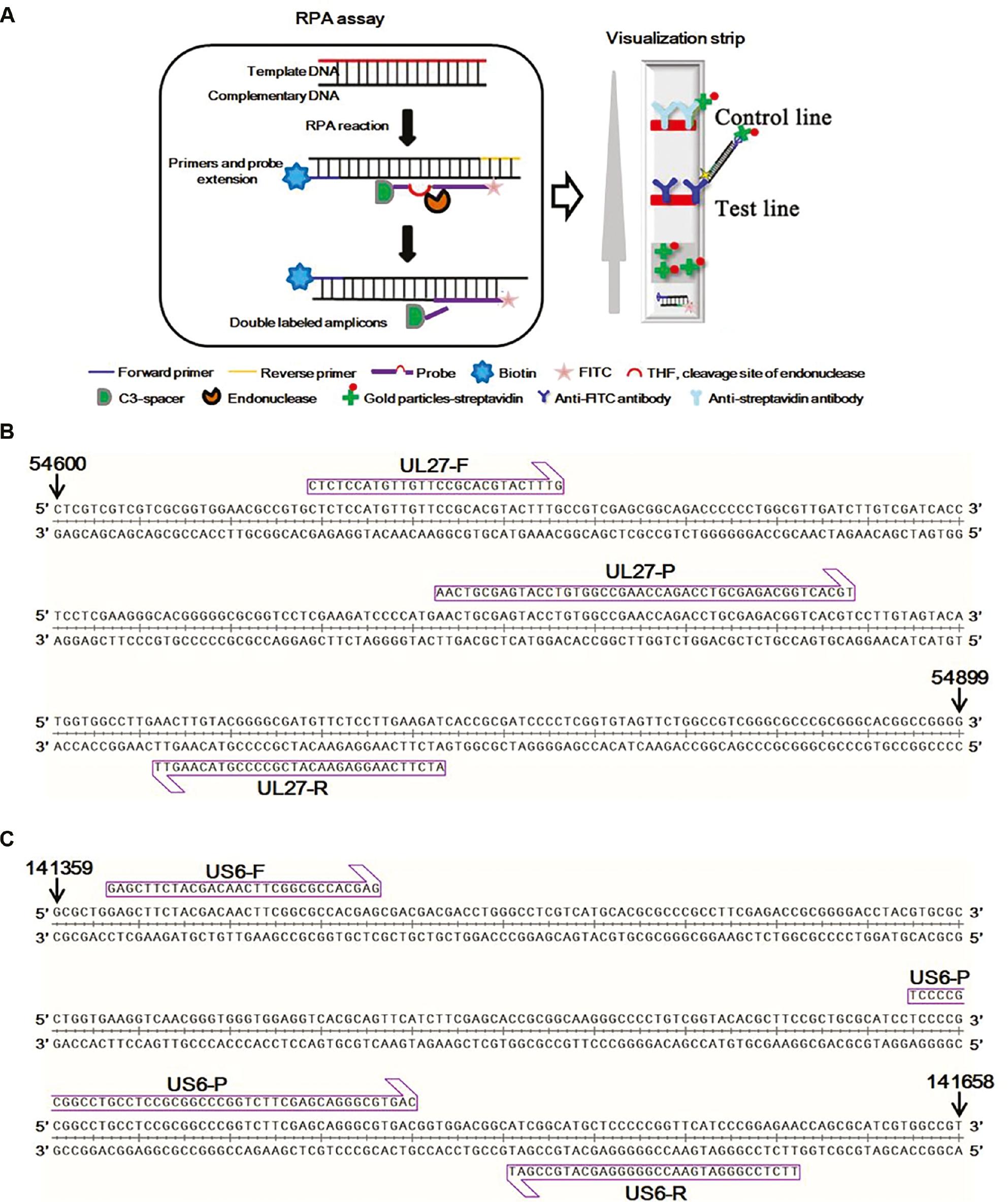
Details of primers and probes used in RPA assays.
(A) Schematic of RPA-VF reaction and vertical flow visualization strip. In short, the recombinase, strand-displacing DNA polymerase and primers initiate the amplification. Then, the special endonuclease cleaves the THF site when the probe hybridizes to the target sequence. As a result, the C3 spacer is dissociated and the probe extension is activated. After amplification, the reaction tube is placed into the closed vertical flow visualization strip device for detecting the RT-RPA products. (B) The target region spanned nucleotides 54,628-54,833 of the complete genome of BV, which is located in the UL27 gene and was used for primer and probe design of RPA-VF-UL27. (C) The target region spanned nucleotides 141,365-141,640 of the complete genome of BV, which is located in the US6 gene and was used for primer and probe design of RPA-VF-US6.
Plasmid construction and DNA standard preparation
Two 500 bp-length sequences of UL27 and US6 (covering target sequences) were synthesized and cloned into the pUC57 vector. The two recombinant plasmids were then prepared and quantified, followed by 10-fold serial dilution. The diluted plasmids were used as standards for subsequent evaluation.
Sensitivity testing
The 2 recombinant plasmids were quantified, then diluted to the indicated concentrations (ranging from 104–100 copies copies/μL). The diluted plasmid standards were used as templates for the analytical sensitivity of the two RPA assays.
Specificity testing
The specificity of the two RT-RPA-VF assays was evaluated by measuring HSV-1, pseudorabies virus (PRV), and infectious bovine rhinotracheitis virus (IBRV) nucleic acids. The nucleic acids were used as templates for amplification under optimum conditions. The plasmid standard and DEPC water were measured as positive and negative controls, respectively.
RESULTS
Primer and probe design
The UL27 and US6 genes of monkey B virus isolate E2490 (GenBank Accession: KY628984.1) were selected for primer and probe design. As shown in Fig 1B, the forward primer (5′-CTCTCCATGTTGTTCCGCACGTACTTTG-3′), reverse primer (5′-biotin-ATCTTCAAGGAGAACATCGCCCCGTACAAGTT-3′), and probe (5′-FITC-AACTGCGAGTACCTGTGGCCGAACCAGACC-THF-GCGAGACGGTCACGT-C3 Spacer-3′) for the UL27 gene were designed to target a 215 bp region. Similarly, a set of primers (forward primer: 5′-GAGCTTCTACGACAACTTCGGCGCCACGAG-3′, reverse primer: 5′-biotin-TTCTCCGGGATGAACCGGGGGAGCATGCCGAT-3′), and a probe (5′-FITC-TCCCCGCGGCCTGCCTCCGCGGCCCGGTCT-THF-CGAGCAGGGCGTGAC-C3 Spacer-3′) was designed to amplify a 276 bp-length region of the US6 gene (Fig 1C). The primers and probe were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China).
Optimization of the two RPA-VF assays
To improve sensitivity, 10-fold dilutions of the synthesized plasmids (pUC57-UL27-Nt and pUC57-US6-Nt) were used as templates for two RPA-VF assays. First, the reactions were performed at different temperatures (33°C, 37°C, 39°C, 42°C, and 45°C) for 30 min. The RPA-VF assay that targets the UL27 gene (RPA-VF-UL27) detected the positive plasmid at a concentration of 1.7×100 copies/μL at 42°C, which was more sensitive than reactions performed at other temperatures (Table 1). The RPA-VF assay that targets the US6 gene (RPA-VF-US6) also reached the best detection level of 1.4×101 copies/μL at 42°C (Table 2). These data suggest that 42°C is the optimal temperature for the 2 assays.
Temperature optimization of the RPA-VF assay targets UL27 gene.
| Temperature (°C) | Template plasmid dilution (copies/μL) | ||||
|---|---|---|---|---|---|
| 1.7×103 | 1.7×102 | 1.7×101 | 1.7×100 | Negative | |
| 33°C | + | + | − | − | − |
| 37°C | + | + | − | − | − |
| 39°C | + | + | + | − | − |
| 42°C | + | + | + | +a | − |
| 45°C | + | + | − | − | − |
aThe result was weakly positive.
Temperature optimization of the RPA-VF assay targets US6 gene.
| Temperature (°C) | Template plasmid dilution (copies/μL) | ||||
|---|---|---|---|---|---|
| 1.4×103 | 1.4×102 | 1.4×101 | 1.4×100 | Negative | |
| 33°C | + | + | − | − | − |
| 37°C | + | + | − | − | − |
| 39°C | + | + | − | − | − |
| 42°C | + | + | + | − | − |
| 45°C | + | + | − | − | − |
To determine the optimal reaction duration, plasmid standards with 10-fold dilutions were used for amplification at 42°C for 20, 30, and 40 min. RPA-VF-UL27 (Table 3) and RPA-VF-US6 (Table 4) reached detection limits when the reactions were ≥ 30 min. Therefore, we selected an amplification time of 30 min as the optimal reaction time for the 2 assays.
Sensitivity comparison
The detection limits of the 2 abovementioned assays were evaluated by using 10-fold-diluted plasmid standards. Under optimal reaction conditions (30 min at 42°C), 3.4 copies of plasmid standards (1.7×100 copies/μL × 2 μL/reaction) were detected by RPA-VF-UL27 (Fig 2A), while RPA-VF-US6 could be performed with a detection limit of 28 plasmid copies (1.4×101 copies/μL × 2 μL/reaction; Fig 2B).

Sensitivity evaluation of RPA assays.
The sensitivity of the RPA assays was evaluated using different concentrations of recombinant plasmids. Ten-fold dilutions of the positive plasmid control were used as templates. The plasmid pUC57-UL27-Nt at different concentrations (ranging from 1.7 × 103–1.7 × 100 copies/μL) was used, and the RPA-VF-UL27 assay was performed at 42°C for 30 min. A negative control was also set, and the result is shown in (A). The plasmid, pUC57-US6-Nt, at different concentrations (ranging from 1.4 × 103–1.4 × 100 copies/μL) was used for evaluation of the RPA-VF-US6 assay (performed at 42°C for 30 min), and the result is shown in (B).
Specificity evaluation
Nucleic acids of several herpesviruses (including HSV-1, PRV, and IBRV) were used for specificity evaluation, while pUC57-UL27-Nt and pUC57-US6-Nt were used as positive controls for the two assays, respectively. The negative control was also set. Negative results were obtained when PRV, IBRV, and control nucleic acids were subjected to RPA-VF-UL27 and RPA-VF-US6 (Fig 3A and B). RPA-VF-UL27 had cross-reactivity with HSV-1 (Fig 3A), which was not observed under RPA-VF-US6 (Fig 3B). Altogether, no false-positive results were obtained for the detection of HSV-1, PRV, and IBRV nucleic acids using RPA-VF-US6, while RPA-VF-UL27 had cross-reactivity with HSV-1 nucleic acids other than herpesviruses nucleic acids.
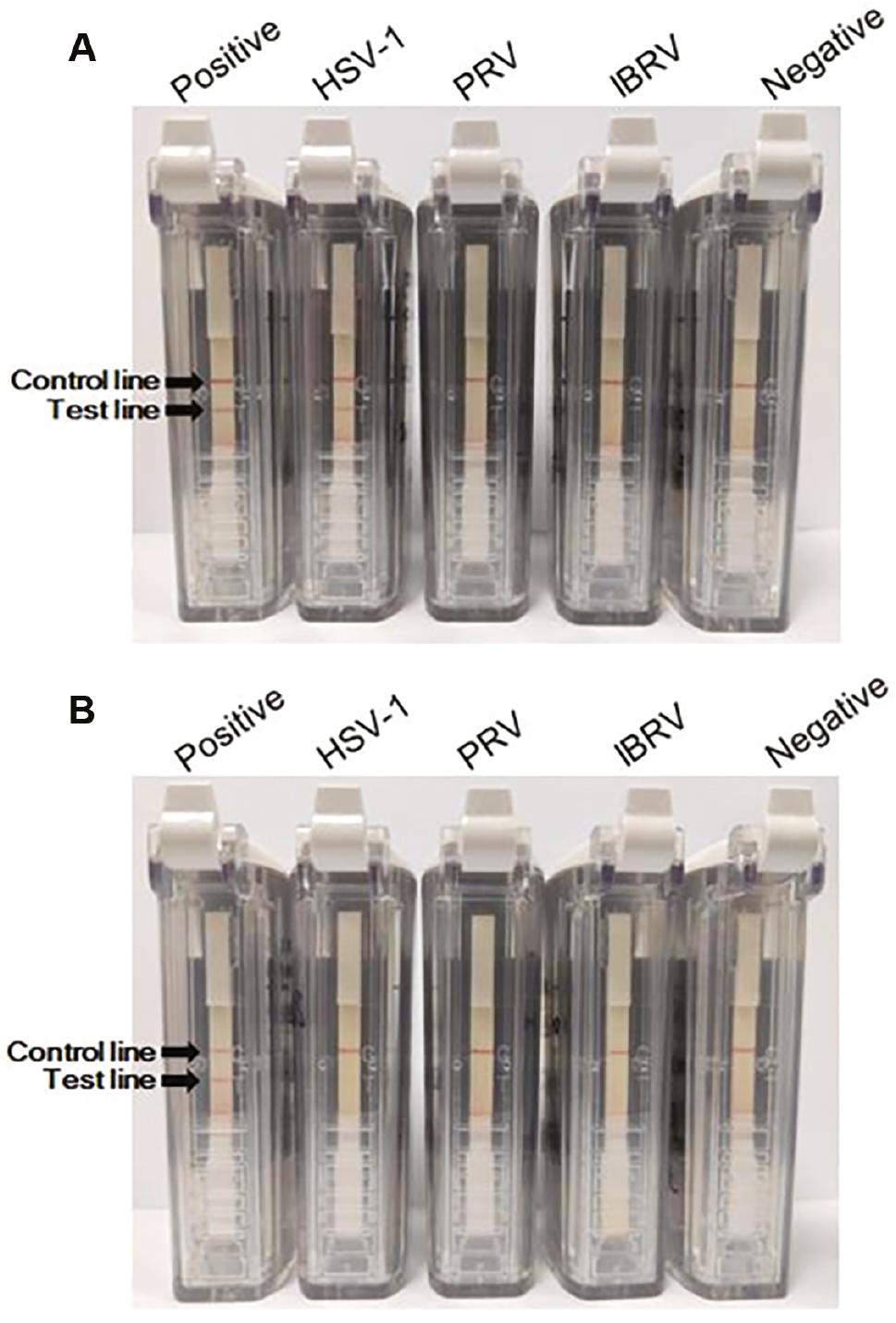
Specificity evaluation of RPA assays.
The specificity of the RPA assays was evaluated using HSV-1, PRV, and IBRV nucleic acids. Recombinant plasmids (pUC57-UL27-Nt and pUC57-US6-Nt) were used as the positive controls of the two assays, while DEPC water was used as the negative control. The specificity evaluation result of the RPA-VF-UL27 assay is shown in (A), and the evaluation result of the RPA-VF-US6 assay specificity is shown in (B).
Room temperature performance
To evaluate the performance of RPA-VF-US6 at room temperature, 10-fold-diluted plasmid standards were used. As shown in Fig 4, even 2,800 copies of plasmid (1.4×103 copies/μL × 2 μL/reaction) were detected at room temperature, indicating the feasibility of the RPA-VF-US6 assay at room temperature.

DISCUSSION
Because of the high human BV infection mortality rate, techniques need to be further developed to improve the diagnostic performance of case detection. It is becoming increasingly important to develop a rapid and reliable method of BV diagnosis to prevent BV transmission.
Isothermal amplification is a novel technique that accomplishes nucleic acid amplification using only a simple temperature controller. This technology has the potential to be applied to point-of-care (POC) testing [21]. Indeed, multiple technologies have been used in the diagnosis of infectious viruses. Cao et al. [22] developed a loop-mediated isothermal amplification (LAMP) assay to detect West Nile virus (WNV) RNA. Navidad et al. [23] developed a rolling circle amplification (RCA)-based approach for porcine circovirus type 2 (PCV2) diagnosis for detection of Chikungunya virus (CHIKV), Telles et al. [24] described a real-time nucleic acid sequence-based amplification (NASBA) method. Song et al. [25] established a visualization assay targeting severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) based on RPA technology under the global COVID-19 epidemic. Moreover, multiple cross displacement amplification (MCDA)- [26] and CRISPR-based [27] detection methods have also been reported for COVID-19 diagnosis.
In this study we applied RPA technology to develop a diagnostic method targeting BV. Compared with reported methods [14,18,19], the entire RPA assay can be performed at a constant temperature without the requirement of sophisticated instruments, such as a PCR apparatus or real-time fluorescent quantitative PCR instrument. By labeling the primer and probe with biotin and FITC, the amplification products are correspondingly labeled. As a result, visual detection of the products is realized through visualization strip analysis. Cross-contamination of amplification products usually results in false-positive test results. To avoid this inherent flaw, we combined a visualization strip with a closed, plastic, leak-proof device. By using the closed VF visualization strip device, the diagnosis result can be visually determined within 2 min, preventing the leakage of amplification products.
Targeting two conserved genes (UL27 and US6), we developed two RPA assays (RPA-VF-UL27 and RPA-VF-US6). Both assays had excellent performance in sensitivity evaluation, and the detection limits of RPA-VF-UL27 and RPA-VF-US6 were 3.4 and 28 plasmid copies, respectively. To obtain a specific assay, we determined whether RPA-VF-UL27 and RPA-VF-US6 cross-reacted with other herpesviruses. The data showed that only the positive plasmid control (rather than HSV-1, PRV, or IBRV nucleic acids) can be detected by RPA-VF-US6, while RPA-VF-UL27 has cross-reactivity with HSV-1.
Because veterinarians, animal care personnels, and laboratory researchers are the main groups at risk for BV infection [8], and the infection of these groups usually occurs through bites, scratches, and other inflicted injuries [6,7], the post-exposure diagnosis of BV is crucial for timely detection of infected cases, which also indicates the importance of POC testing of BV infection. As a result, quick diagnosis is suitable for detection in the field laboratory. To evaluate the practicability of our RPA assay, we determined the amplification performance of RPA-VF-US6 at room temperature, and the results showed that even 2,800 copies of plasmid could be detected, suggesting the potential of RPA-VF-US6 in POC testing. In addition, the use of a closed, plastic device not only realizes visual readout of the detection result, but also reduces the leakage possibility of amplification products.
In summary, we developed two RPA assays for BV visualization diagnosis. Because of its sensitivity and specificity, RPA-VF-US6 was selected for further evaluation. Even 2,800 plasmid copies could be detected at room temperature. In addition, the use of a closed VF visualization strip device not only saves time in result readout, but also reduces the possibility of contamination by amplification products. Taken together, RPA-VF-US6 has multiple advantages and is especially suitable for POC testing in field laboratories.