
INTRODUCTION
Ticks are obligate blood-sucking ectoparasites of many vertebrate animals and important vectors for various zoonotic diseases [1]. Emerging tick-borne pathogens have raised a global concern. Ticks are globally distributed and can adapt to various environments and hosts [2]. Ticks have a life cycle from ‘on-host’ feeding to ‘off-host’ free-living, thus they are challenged by numerous microorganisms [3–5]. Therefore, it is an important prerequisite to characterize the microbial community of ticks and to reveal novel insights for the strategic control of ticks and tick-borne diseases in the future [4,6,7]. High-throughput sequencing has provided insight into the microbial diversity associated with ticks [8–11]. The 16S rRNA full-length sequencing is a high-throughput technique that can be used to detect non-culturable microbes and reveal the entire population of tick-borne microbes [5].
In this study tick samples were obtained in Yunnan, China. Combined with tropical humidity, the special topographic range maintains extremely high biodiversity and high degrees of endemism. Recently, several surveys have reported many tick-borne pathogens in Yunnan province, such as Babesia microti, Ehrlichia chaffeensis, Coxiella burnetii, and Jingmen tick virus [12–20]. This work aimed to clarify the microbial populations, including pathogens, associated with ticks using 16S rRNA full-length sequencing technology, and PCR amplification and sequencing to identify the diversity of tick-borne microbiota in this region.
METHODS
Locations and collection of ticks
Ticks were collected during the 2015 and 2016 spring and autumn intervals by dragging a white fabric flag over vegetation or detaching from animals in GengMa, Tengchong, Lanping, Weixi, and Deqin counties (Yunnan Province, southern China; Fig 1). Tick species were identified by morphology and confirmed by16S rRNA (rrs) analysis [13]. All tick specimens were stored alive and separately in sterile tubes at 4–6 °C. Ticks were carefully surface-sterilized under stringent conditions in a biohazard cabinet. The ticks were first submerged in 3 % H2O2, followed by 70 % EtOH for surface sterilization.

DNA extraction
Air-dried ticks were homogenized in sterile glass micro-blenders using liquid nitrogen, then the genomic DNA for constructing eubacterial 16S rRNA gene libraries were extracted. DNA was isolated using the DNeasy Tissue Kit (Qiagen, city, Germany) according to the manufacturer’s instructions from individual field-collected adults. DNA concentration and quality were determined by spectrophotometry using the NanoDrop 2000c and agarose gel electrophoresis. The following 16s rRNA sequencing was performed by the Novogene company (Beijing, China).
16S rRNA full-length sequencing
The pooled tick DNA samples were used for the 16S rRNA full-length sequencing. The eubacterial 16S rRNA gene was constructed by amplifying an approximately 1500-bp fragment of a 16S rRNA gene using eubacterial universal primers (27 F/1492R) [21] with specific barcodes. PCR was carried out in a 25-μl final volume mixture using 2 μl of the extracted template DNA with the following cycling parameters: initial denaturation at 94°C for 10 min; 30 cycles at 94°C for 40 s, 55°C for 1 min, and 72°C for 1 min; with a final extension at 72°C for 10 min. Post-amplification quality control was performed using a bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
SMRTbell libraries were prepared from the amplified DNA by blunt ligation, according to the manufacturer’s instructions (Pacific Biosciences, city, state, country). Purified SMRTbell libraries were sequenced on a single PacBio Sequel cell (Novogene, city, China).
Microbiome analysis
Raw fastq files were de-multiplexed and quality-filtered using QIIME (v1.8.0) with previously described criteria [22], and operational taxonomic units (OTUs) were clustered with a 97% similarity cut-off using UPARSE (version 7.1; http://drive5.com/uparse/). Alpha and beta diversities were calculated based on the normalized-OUT abundance information, which was obtained using the sample with the fewest sequences as a standard. Samples were first clustered according to beta-diversity matrices, followed by an unweighted pair group method with arithmetic mean (UPGMA) clustering based on the UniFrac distance matrix [14]. Differences in alpha diversity metrics between groups were assessed with the Kruskal-Walli’s test (p<0.05) using QIIME, and bacterial beta diversity was compared between groups with the PERMANOVA test (p<0.05) on QIIME. We compared the representative OTU sequence with the Silva database used Blastn to define the taxonomy, and the best match with an identity > 97% were used to assign the OTU taxonomy.
PCR amplification
Conventional PCR methods were used to detect and determine the tick-borne pathogenic genera, including Anaplasma, Ehrlichia, Neoehrlichia, Rickettsia, Borrelia, and Babesia. The nested PCR for the Anaplasmataceae-specific 16S rRNA gene with primers (Eh-out1/Eh-out2 and Eh-gs1/Eh-gs2), followed by sequencing was used to amplifyy Anaplasma, Ehrlichia, and Neoehrlichia [15]. Nested PCR with primers (23S3/23Sa and 23S5/23S6) of the Borrelia 5S-23S rRNA intergenic spacer gene were used to identify Borrelia [16]. A PCR targeting the partial 18S rRNA gene of Babesia species, including piroplasm, with primers (PIRO-A and PIRO-B) was used to identify Babesia [23].
We first screened each sample using the nested PCR for the Anaplasmataceae-specific rrs gene [15] and Rickettsiae-specific citrate synthase gene (gltA) to determine the diversity of Rickettsia species in the samples [24]. The nearly entire rrs gene sequences (1420bp), gltA (1232bp), 190-kDa outer membrane protein coding gene (ompA [625bp]), ompB (435bp), surface cell antigen (sca4 [817bp]), sca1 (419bp), and 17-KDa protein (htrA [503bp]) of the Rickettsiae-positive species were further amplified. Amplicons were extracted from 2% agarose gels and purified using a QIAquick Gel Extraction Kit (Qiagen, Inc., Valencia, CA, USA), according to the manufacturer’s instructions. All amplifications were confirmed by Sanger sequencing.
Phylogenetic analyses
The obtained sequences were compared with the sequences available in GenBank using BLAST (http://blast.ncbi.nim.nih.gov/Blast.cgi). Phylogenetic trees were constructed via Mega 5.0 [25] using the neighbor-joining method with the Kimura two-parameter model, and bootstrapping was performed with 1000 replicates.
RESULTS
A total of 191 adult ticks (153 females and 38 males) were collected, among which 103 were host-seeking ticks and 88 were engorged ticks from animals. The tick species included Ixodes ovatus (82) I. acutitarsus (7), I. granulatus (30), Rhipicephalus microplus (22), and Haemaphysalis kolonini (50), which were identified by morphology and 16S rRNA analysis. H. kolonini was a new species in the subgenus, Alloceraea Schulze (Ixodidae: Haemaphysalis), in China [26].
16S rRNA full-length sequencing
The 16S rRNA full-length sequencing was performed on the following eight pooled samples: I. ovatus from Lanping (LPLAX); I. ovatus from Gengma (GMLAX); I. acutitarsus from Gengma (GMRF); I. granulatus from Tengchong (TCLX); I. ovatus from Tengchong (TCLAX); R. microplus from Tengchong (TCWX); H. kolonini from Weixi (WXKLN); and H. kolonini from Deqin (DQNBE; Table 1).
Sampling details and PCR amplification of tick-borne pathogenic genera.
| Sample name | Tick species | Hosts | Sampling location | No. of ticks | Anaplasma | Ehrlichia | Candidatus Neoehrlichia | Rickettsia | Borrelia | Babesia |
|---|---|---|---|---|---|---|---|---|---|---|
| LPLAX | Ixodes ovatus | Free | Lanping, Yunnan | 29 | 5 | 6 | 4 | 4 | ||
| GMLAX | Ixodes ovatus | Free | Gengma, Yunnan | 25 | 8 | 1 | 2 | 13 | 1 | |
| GMRF | Ixodes acutitarsus | Free | Gengma, Yunnan | 7 | 6 | |||||
| TCLX | Ixodes granulatus | Mouse | Tengchong, Yunnan | 30 | 2 | 6 | 10 | 18 | 7 | |
| TCLAX | Ixodes ovatus | Sheep | Tengchong, Yunnan | 28 | 4 | 12 | 8 | |||
| TCWX | Rhipicephalus microplus | Cows | Tengchong, Yunnan | 22 | 14 | 9 | 4 | |||
| WXKLN | Haemaphysalis kolonini | Free | Weixi, Yunnan | 42 | 5 | 2 | 31 | 6 | ||
| DQNBE | Haemaphysalis kolonini | Cows | Deqin, Yunnan | 88 | 8 | 3 | 6 |
A total of 135,168,306 quality reads for 8 samples were obtained with an average read length of 1481 bp. The data were uploaded into QIIME, and the analysis revealed 10,692 valid OTUs with 1060 OTUs coming from TCLAX, 1488 from TCWX, 1148 from WXKLN, 1138 from DQNBE, 1551 from LPLAX, 1353 from GMLAX, 1225 from GMRF, and 1729 from TCLX.
Taxonomic abundance of the obtained sequences was summarized at the phylum and genus levels; 11 phyla were identified in total from all samples. Among these identified microbes, Proteobacteria were the most abundant in all samples except TCLX, followed by Firmicutes, while Tenericutes was the most abundant in TCLX (89.34%), followed by Proteobacteria (10.28%). Actinobacteria was also abundant in TCWX (2.38%), LPLAX (8346%), and GMLAX (2.32%), and Deinococcus accounted for 2.95% in LPLAX and 10.84% in GMRF (Fig 2(A)).
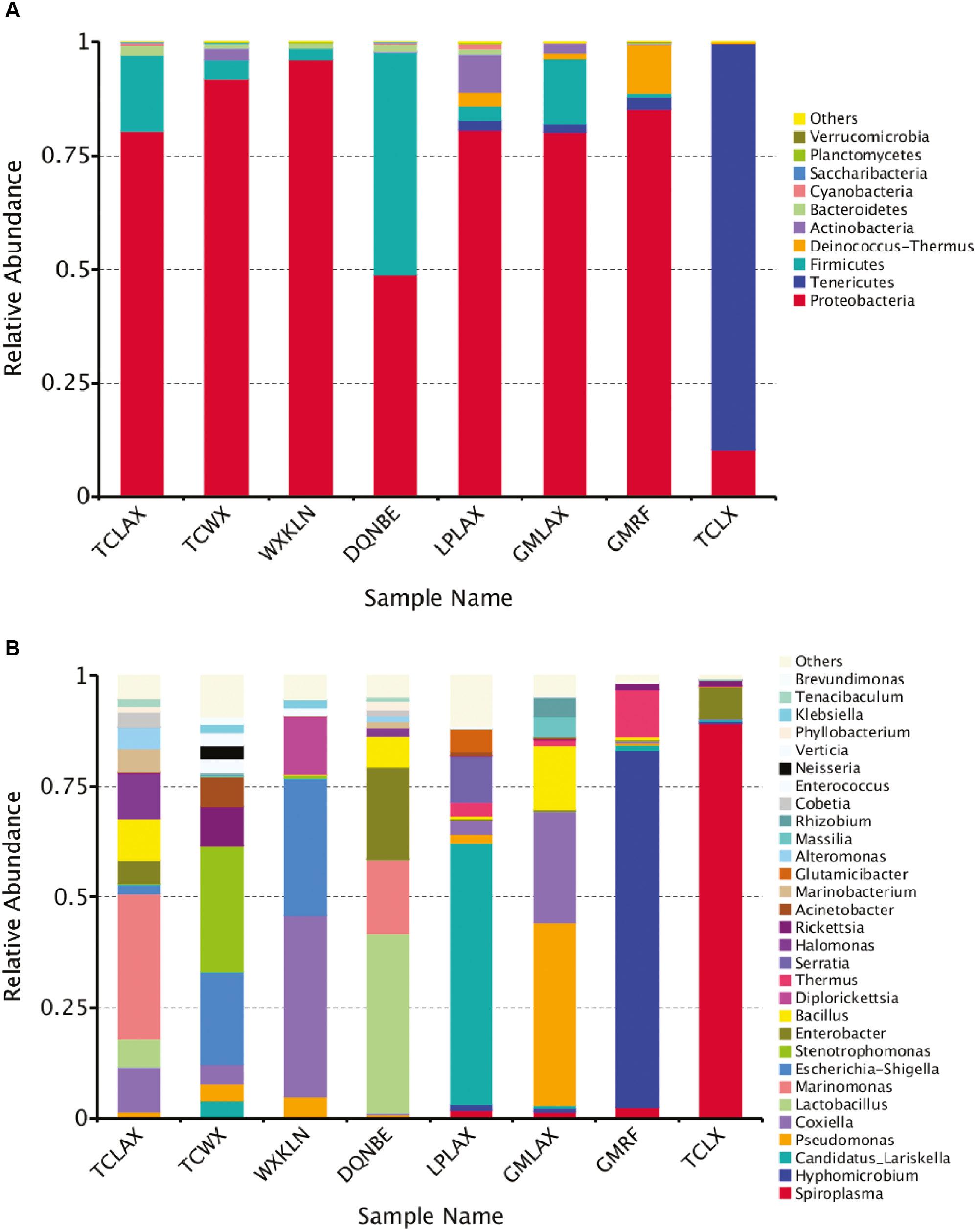
A: Relative abundance of the characterized microbes at the phylum level. B: Relative abundance of the characterized microbes at the genera level.
One hundred twenty-six genera were identified from all the samples. Marinomonas (32.71%) was the most abundant genus in TCLAX, followed by Coxiella (10.21%). Stenotrophomonas (28.45%) occupied the most populations in TCWX, followed by Escherichia (20.80%) of the genera. Coxiella accounted for 40.79% and Escherichia accounted for 31.20% of the genera in WXKLN. Lactobacillus and Enterobacter accounted for 40.41% and 20.74% of the genera in DONBE, respectively. Candidatus Lariskella (59.02%) was the most abundant genus in LAPLAX, followed by Serratia (10.53%). Pseudomonas and Coxiella accounted for 41.29% and 25.19% in GMLAX, respectively. Hyphomicrobium and Thermus accounted for 80.58% and 10.84% of the genera in GMRF, respectively. Spiroplasma (89.34%) was the most abundant genus in TCLX, followed by Enterobacter [7.08%; Fig 2(B)].
Alpha diversity
Alpha diversity was accessed using rarefaction curves (S1 Fig). The rarefaction curves revealed that all sample sequencings were almost close to the saturation plateau, which indicated that the sequencing depth was representative of the microbial communities in each sample.
The number of assigned OTU are shown in S2(A) Fig based on the taxonomy. The majority of species were in TCWX, while the number of assigned OTUs was the least in TCLX, indicating that TCWX had the highest overall bacterial diversity and TCLX had the lowest. There were seldom common OTUs among the samples from the same locations [Tengchong and Gengma; S2(B) Fig]. In addition, I. ovatus showed that there were only three shared OTUs between Tengchong and Gengma, and eight between Gengma and Laping. The microbial diversity was much different independent of the geographic distribution or tick species.
Beta Diversity
Beta diversity (genetic relatedness) was calculated using unweighted UniFrac. The principal coordinate analysis (PCoA) plot is shown in Fig 3. There were three possible clusters among all samples; the microbial composition of GMRF and GMLAX from the same regions were more similar than other samples. The three I. ovatus ticks were spread out on graphs of the first principal coordinate (PC1).

The two values above and below represent Weighted Unifrac and Unweighted Unifrac distance, respectively, as shown in the heatmap (S3 Fig). Generally, each sample was highly diverse from each other. It was shown that the difference between DNQBE and TCLAX was the smallest (0.738/0.600), while WXKLN was significantly different from TCLX (1.769/0.867).
PCR amplification
We individually identified Anaplasma, Ehrlichia, Candidatus Neoehrlichia, Rickettsia, Borrelia, and Babesia in our tick samples (Table 1). This result suggested that ticks from Yunnan carried various tick-borne pathogens. In addition, there was a significant difference in the positive rate of tick-borne pathogens in this region.
Because Rickettsia was diverse in this region, we further sequenced Rickettsia species in all individual samples. Rickettsia sp. YN01 was detected in 4 I. granulatus (13.3% [4/30]) in TXLX (Table S1). The sequences of rrs, gltA, ompA, ompB, and sca4 were genetically most similar to Rickettsia sp. IG-1 (99.4%, 99.9%, 100%, 99.5%, and 99.7% similarity) with 8 bp, 1 bp, 0 bp, 2 bp, and 2 bp differences, respectively (Table 2). The 17 KDa sequence was not available for Rickettsia sp. IG-1, and our sequence showed 5 bp differences (99% identity) with R. rickettsii (CP006010). Rickettsia sp. YN02 infection occurred in 2 (28.6%) I. acutitarsus in GMRF (Table S1). Of the rrs, gltA, ompA, ompB, and sca4 sequences, 17KDa showed the highest identity with R. raoultii (99.5%, 99.7%, 98.0%, 98.0%, 98.4%, and 98.6% similarity) and had 6bp, 3bp, 10bp, 8bp, 13bp, and 8bp differences, respectively (Table 2). Rickettsia sp. YN03 infected 4 (14.3%) I. ovatus in TCLAX and 14 (63.6%) Rhipicephalus microplus in TCWX (Table S1). Of the rrs, gltA, ompA, ompB, sca4, and sca1 sequences, 17KDa shared 99.4%, 99.5%, 95.1%, 99.0%, 98.0%, 99.2%, and 98.9% similarity to R. heilongjiangensis with 8 bp, 6 bp, 25 bp, 4 bp, 16 bp, 3 bp, and 4 bp differences, respectively (Table 2). Phylogenetic analysis of 4670 bp concatenated sequences Of the 4670 concatenated sequences of rrs, gltA, ompB, and sca4 genes and phylogenetic analysis of ompA, the 17 KD gene suggested that R. sp. YN01 was a variant strain of Rickettsia spp. IG-1, but Rickettsia sp. YN02 and Rickettsia sp. YN03 were two potentially new SFGR species (Fig 4).
Maximum identities of spotted fever group rickettsial sequences obtained from ticks with validated Rickettsia spp. published in GenBank, Yunnan, China.
| Genes | Sequence length (bp) | Length of ORF to R. rickettsii | Rickettsia sp. YN01 | Rickettsia sp. YN02 | Rickettsia sp. YN03 | Primer | Nucleotide sequence (5′-3′) |
|---|---|---|---|---|---|---|---|
| rrs | 1390 | 60-1448 | 99.4% to Rickettsia spp. IG-1(1382/1390 EF589608) | 99.5% to Rickettsia raoultii (1383/1389 CP010969) | 99.4% to Rickettsia heilongjiangensis (1381/1389 CP002912) | Eh-out1 Eh-3-17 Eh-out1 Eh-out2 | TTGAGAGTTTGATCCTGGCTCAGAACG TAAGGTGGTAATCCAGC TTGAGAGTTTGATCCTGGCTCAGAACG CACCTCTACACTAGGAATTCCGCTATC |
| gltA | 1220 | 32-1249 | 99.9% to Rickettsia spp. IG-1(1194/1195 EF219460) | 99.7% to Rickettsia raoultii (1216/1219 CP010969) | 99.5% to Rickettsia heilongjiangensis (1214/1220 CP002912) | CS2d CSEndr RpCS877 RpCS1258 | ATGACCAATGAAAATAATAAT CTTATACTCTCTATGTACA GGGGACCTGCTCACGGCGG ATTGCAAAAAGTACAGTGAACA |
| ompA | 510 | 6229-6738 | 100% to Rickettsia spp. IG-1(501/501 EF219466) | 98.0% to Rickettsia raoultii (497/507 CP010969) | 95.1% to Rickettsia heilongjiangensis (483/508 CP002912) | Rr190.70f Rr190.602r | ATGGCGAATATTTCTCCAAAA AGTGCAGCATTCGCTCCCCCT |
| ompB | 407 | 844-1249 | 99.5% to Rickettsia spp. IG-1(405/407 EF219461) | 98.0% to Rickettsia raoultii (399/407 CP010969) | 99.0% to Rickettsia heilongjiangensis (393/397 CP002912) | rompB OF rompB OR rompB SFG IF rompB SFG/TG IR | GTAACCGGAAGTAATCGTTTCGTAA GCTTTATAACCAGCTAAACCACC GTTTAATACGTGCTGCTAACCAA GGTTTGGCCCATATACCATAAG |
| sca4 | 814 | 1790-2603 | 99.7% to Rickettsia spp. IG-1(812/814 EF219462) | 98.4% to Rickettsia raoultii (801/814 CP010969) | 98.0% to Rickettsia heilongjiangensis (794/810 CP002912) | sc4-1 Rj2837r sc4-3 sc4-4 | ATGTCTCTGAATTAAGCAATGC CCTGATACTACCCTTACATC AATTATTAGGCTCTGTATTAAAGA GAAAGGATAGCACGAAAAGTA |

Phylogenetic analyses based on A: concatenated datasets of rrs, gltA, ompB, and sca4 genes sequences (4670 bp). The GenBank accession numbers were as follows: KY411132, KY411134, KY469279, and KY411136 (Rickettsia sp. YN01); KY411133, KY411135, KY469278, and KY411137 (Rickettsia sp. YN02); KY433580, KY433588, KY433585, and KY433587 (Rickettsia sp. YN03). B: Nucleotide sequences of the 17-kDa protein-encoding (407 bp). C: Outer member protein A (494bp). Boldface indicates the newly discovered Rickettsia genotype (Rickettsia sp. YN01, Rickettsia sp. YN02, Rickettsia sp. YN03).
DISCUSSION
This study aimed to assess and compare the diversity of bacterial populations of I. ovatus, I. acutitarsus, I. granulatus, R. microplus, and H. kolonini. We identified 126 different genera, including pathogenic genera, such as Anaplasma, Ehrlichia, Candidatus Neoehrlichia, Borrelia, and Babesia. Further identification using species-specific PCR clarified three different tick-borne rickettsiae (Ricekttsia. sp YN01, Ricekttsia. sp YN02, and Ricekttsia. sp YN03) in the ticks used in this study.
A total of 11 phyla were detected in all samples, and Proteobacteria was dominant in I. ovatus, I. acutitarsus, R. microplus, and H. kolonini, which is consistent with the composition of microbiota in many other tick species, except I. granulatus, in which Tenericutes was the most abundant. The second dominant phylum, Firmicutes, was abundant in I. ovatus, I. acutitarsus, R. microplus, and H. kolonini, which was also previously reported in H. tibetensis and I. scapularis. Recently, a better statistic method to calculate the differences in taxonomic abundance (the ANOVA-like differential expression [ALDEx2] package) has been implemented. The advantage is that all features have a one-to-one transformation, thus allowing observation of all changes [27].
The results from the PCA sequences indicated that there were differences in the microbial populations of ticks. The samples from the Gengma were clustered together, which seemed to have a more similar microbial composition than others. Ticks can obtain microorganisms through a variety of ways, such as transovarial transmission, and from the environment, host animals during blood feeding, and mating partners. Bacterial community in these samples were highly diverse, which might be caused by different detached hosts and diverse ecosystems in Yunnan province.
Rickettsia sp. IG-1 was first isolated from I. granulatus in Taiwan [28] and a similar strain was also identified in the same tick species in Japan [29]. We identified a variant strain of Rickettsia sp. IG-1 with an 8-bp difference (1382/1390) of the rrs gene in I. granulatus. Thus, I. granulatus appears to be the vector of this new Rickettsia species.
Yunnan is internationally known as a “Global Biodiversity Hotspot.” The diverse climate, different altitudes, and complicated topography produce a wide range of flora and fauna. Yunnan also borders Vietnam, Laos, and Myanmar. The rich natural environment and frequent traveler movement around the border make the zoonosis a great local public health burden. I. granulatus, I. ovatus, and I. acutitarsus are all recognized as the representative Ixodes species in southeast Asia, and human biting activities have been observed [30,31]. I. granulatus and I. ovatus also harbor genetic diversity of Borrelia spirochetes [32,33], while I. granulatus is recognized as the vector of R. honei [34], and I. ovatus is thought to transmit B. microti [35] and tick-borne encephalitis [36]. R. microplus has a wide global distribution range and is a vector of many zoonotic diseases because R. microplus feeds on many animal hosts. We identified a new tick species, H. kolonini, which carried various species of Anplasma, Borrelia, and Rickettsia in this study. In addition, our study of microbiota in the ticks revealed the complexity of ecologic interactions between host and microbe and provided insights for the biological control of ticks.