
Human plague, caused by Yersinia pestis, a bacterium which first and foremost occurs in wildlife rodent species but occasionally spills over to people: hence the title “Yersinia pestis and Plague”. Plague has brought about three pandemics in history, including the first pandemic Justinian plague (from around 541 CE), the second Black Death (from around 1347 CE), and the third modern plague (from around 1880 CE). Since the etiologic agent for plague was identified by Dr. Alexander Yersin during the third pandemic in Hong Kong of China in 1894 [1], more than 120 years have passed, and we have a better understanding of the physiology, pathogenesis, and evolution of Y. pestis. We have developed technologies for plague diagnosis and treatment. Plague is effectively controlled in most endemic regions, except some parts of Africa. The wide distribution of natural plague foci in Asia, Eurasia, Africa, and the Americas, with occasional occurrences in both rodents and humans or following long-distance travel of plague-infected individuals into large cities reminds us of the threat of a plague outbreak. In this review, we will summarize what we know and what we do not know about plague and its causative agent, Y. pestis. The key issues that need to be resolved are also included (Box).
Key issues to be solved for Yersinia pestis and plague
Improved understanding of sensing and adapting of Y. pestis to temperature shifts during its invasion from flea to animals;
Improved understanding of survival of Y. pestis in host innate immune cells during the early stage of infection;
Improved understanding of heavy growth of Y. pestis in host blood by potent immuno-suppression for flea transmission;
Improved understanding of the driving force shaping Y. pestis’ ancestry and the detailed dynamics during its evolution;
Improved understanding of ecological interactions of Y. pestis, the hosts and natural environments for long-term survival of natural plague foci;
Improved understanding of molecular mechanisms behind complex interaction between Y. pestis and fleas;
Improved understanding of molecular mechanisms of early interactions between Y. pestis and the host cells;
Development of on-site diagnostic techniques and precision treatment strategies;
Development of effective and safe vaccine for long-term protection;
Effective preparedness for Y. pestis bioterrorism.
KNOWN 1: PHYSIOLOGY OF Y. PESTIS
Growth properties and cellular structures
Y. pestis is a Gram-negative, non-sporulating and non-motile coccobacillus that grows in broth culture. Y. pestis often exhibits bipolar staining with Giemsa or Wayson dye. Y. pestis grows on a variety of media under aerobic or facultatively anaerobic conditions at temperatures between 4°C and 40°C (optimum, 26°C −28°C) and can survive within a pH range from 5.0-9.6 (optimum, approximately 7.5) [2].
Y. pestis typically grows as grey-white, translucent, non-hemolytic colonies within 24 h on blood or chocolate agar, and as yellow and opaque colonies with an irregular, ‘fried egg,’ shiny appearance after 48 h. In some, but not all broth cultures, Y. pestis aggregates with flocs typically attached to the sides of the tube, often projecting a stalactite shape and leaving a clear broth at 48 h [3].
Y. pestis has typical cell structures and antigen compositions, like other enteric bacteria, and produces a short-chain, rough lipopolysaccharide (referred to as lipo-oligosaccharide) that lacks the O antigen due to the absence of some genes from the O-antigen cluster [4]. This unique feature might be an evolution-associated adaptation that contributes to systemic infectivity by this pathogen. Y. pestis lacks a true capsule, but frequently produces a unique glycoprotein envelope known as the surface capsule at temperature above 33°C [5].
Genomic composition
The deadly pathogen, Y. pestis, is a clone that emerged from the self-limiting gastroenteric pathogen, Y. pseudotuberculosis [6–9]. Approximately 97% similarity is shared at the chromosomal DNA level [6], and like other pathogenic Yersiniae, Y. pestis contains the plasmid, pCD1 (70-75 kb) [10]. During evolution, Y. pestis acquired two additional plasmids horizontally, pMT1 (100-110 kb) and pPCP1 (9.5 kb), and a high pathogenicity island consisting of 32 chromosomal genes that is unique to Y. pestis. Some determinants encoded by the plasmids, pMT1 and pPCP1, facilitate Y. pestis-specific tissue invasion, survival in flea vectors, or possibly heavy growth in host blood [11–15].
Gene modification and loss are attributed to modifications of cellular structural or regulatory networks or elimination of activities no longer required for the Y. pestis new lifecycle [16]. For example, mutation or interruption of yadA, inv, and ail which encode adhesin or invasin attenuates the activities usually attributed to enteropathogenic virulence [17–19]. Urease is essential for pathogenesis in other members of Yersinia spp. (Y. enterocolitica and Y. pseudotuberculosis) [20], but Y. pestis does not exhibit ureolytic activity due to a premature stop codon of the ureD gene [21]. Even though the ure operon is transcriptionally upregulated at 26°C, this locus might not be directly involved in the Y. pestis life cycle, but likely reduces the toxicity to fleas, thus increasing the probability of maintaining an infection in a larger population [22].
Nutrition & metabolism traits
Almost all Y. pestis strains require media supplementation with isoleucine, valine, methionine, phenylalanine, and glycine or threonine for successful growth; however, the nutrient requirements differ depending on the temperature. Additional nutrients, such as biotin, thiamine, pantothenate, and glutamate, are required at 37°C.
Y. pestis strains have different abilities to ferment glycerol and arabinose, and reduce nitrate. Based on this finding, Y. pestis strains have been classified into five biovars (Antiqua, Mediaevalis, Orientalis, Microtus, and Intermedium) [23,24]. The strains of the former three biovars are highly virulent in animals and humans; however, Microtus, including ‘pestoides,’ is non-virulent or opportunistic in larger mammals, but virulent in small rodents [25].
Y. pestis has a complete Embden-Meyerhof pathway, but no functional pentose-phosphate pathway due to missense mutations in the glucose 6-phosphate dehydrogenase-encoding gene zwf. Unlike Y. pseudotuberculosis, the glyoxylate bypass pathway is constitutively-expressed in Y. pestis due to de-repression from the IclR transcriptional repressor [26], which may explain the ability of Y. pestis to metabolize acetate and fatty acids provided by the host.
UNKNOWN 1: REGULATION OF Y. PESTIS PHYSIOLOGY IN VITRO AND IN VIVO
In contrast to the ancestor of Y. pestis, Y. pseudotuberculosis, a self-limiting gastroenteric pathogen, Y. pestis has evolved to be a deadly pathogen occupying different niches [6,7]. Y. pestis only circulates within a narrow host range between rodent reservoir hosts and flea vectors in natural settings. The first challenge for Y. pestis survival in its lifecycle is sensing and adapting to temperature shifts while avoiding host innate immune cells during the early stage of infection and in host blood after release from innate immune cells, including macrophages, [27] is problematic as Y. pestis develops into a systemic infection. During the complex lifestyle of Y. pestis, the intense or even life-threatening environmental changes are concomitant with a series of dynamic regulatory physiologic responses. Of note, we are still far from understanding Y. pestis physiology and pathogenesis at the transcriptional and post-transcriptional level [28].
KEY PHYSIOLOGIC TRAITS
Temperature sensing
As an alternative, Y. pestis grows in fleas or in warm-blood mammalian hosts during its lifecycle. Fleas are often infected with Y. pestis by sucking blood of a bacteremic mammal. The bacteria form a biofilm mass in the flea foregut at temperatures below 25°C [29]. Y. pestis distinguishes the temperature shift between environmental and body temperatures of hosts during the transmission process. Interestingly, most of the putative virulence factors are transcriptionally-regulated by temperature shifts and are active at 26°C or 37°C. Under the control of a fourU RNA thermometer switch [30], the histone-like regulator, YmoA, negatively modulates the virulence effectors of the type III secretion system (T3SS) through the transcriptional activator, LcrF [31]. This thermos-sensing pattern is quickly responsive and energy-efficient to match the changing temperature environments encountered by Y. pestis.
Low-calcium response
The low-calcium response (LCR) in Y. pestis can be induced in vitro under low Ca2+ or Ca2+-free conditions at 37°C, but not 26°C, in which growth cessation is coordinated with upregulating the T3SS [32]. Triggering the expression and secretion function of the T3SS is used to stimulate the in vivo signal that occurs when Y. pestis contacts host cells during infection [33]; however, growth cessation might be an artifact that only occurs in vitro. Expression of the T3SS components encoded on pCD1 are shared among all three pathogenic Yersiniae and initiate secretion of Yersinia outer proteins (Yops) into the host cell cytoplasm, which prevents macrophage phagocytosis and inhibits the host immune response [34].
Survival within macrophages
Survival inside macrophage vacuoles is considered critical in the early stages of the Y. pestis life cycle within warm-blooded hosts [35–37]. Y. pestis isolated from fleas has been reported to exhibit resistance to phagocytosis [38] and the host innate immune cells demonstrate different responses to flea-transmitted and needle-inoculated Y. pestis [39]. These findings serve as reminders that we must be prudent in extrapolating the experimental results from needle-injected challenge of laboratory animals to detail the process of natural infections through flea feeding. The intracellular microenvironment of macrophages may provide a temporary shelter for the organism and induce the synthesis of anti-phagocytosis factors for subsequent release into the extracellular environment [40]. PhoP/PhoQ, a two-component regulatory system, is likely important for Y. pestis survival within macrophages via pleiotrophic effects on gene expression [41].
Counteraction of biometal sequestration
During Y. pestis infection, the host microenvironments are thought to sequester key biometals, such as iron, zinc, and manganese. Y. pestis utilizes an iron-scavenging siderophore, Yersiniabactin (Ybt), and iron transporters (Yfe and Feo) to overcome iron deprivation [42–45]. In addition, the zinc transporter, ZnuABC Ybt siderophore, also contributes to zinc acquisition as the second zinc transporters in mammalian and flea hosts during infection [45–47]. Two Mn transporters, Yfe and MntH, that are functional in Y. pestis have important roles in bubonic plague progression [48].
Regulation of the physiologic stress response in vitro and in vivo
A complex lifestyle requires Y. pestis to monitor environmental cues and regulate stress-responses accordingly to ensure environmental adaptation in hosts or vectors.
The stressful conditions in vitro stimulating intracellular or external microenvironments encountered by Y. pestis during infection and the life cycle have been used in studies on regulation of stress responses. Research on stimulons (temperature, osmolarity, ion, oxidative, acid, and nutrition) and regulons (Fur, PhoP, OmpR, and OxyR) refines the regulatory responses and identifies the differentially-regulated genes that are important for Y. pestis physiology and pathogenesis [49]. An integrated analysis showed that expression of the putative virulence locus in Y. pestis (hms, caf1, T3SS, and psa) is responsive to a wide range of environmental stresses and multiple regulatory proteins. Other genes responsible for cellular metabolism are also active upon exposure to multiple stresses, including energy metabolism, sulfur metabolism, ribosome protein biosynthesis, iron uptake, heme synthesis, and utilization to chemotaxis and motility [49].
Virulence-associated genes or loci, such as the T3SS, pgm locus, pH6 antigen and pla encoded by pPCP1, are regulated in established bubonic or pneumonia plague. Iron deprivation and nitric oxide (NO)-induced stress are thought to be more reactive based on regulation of corresponding pathways in the rat bubo [50]. The genes associated with antioxidant stresses are strongly induced in intracellular Y. pestis [51]. In a flea transmission model, regulation of genes involved in innate immunity and pathogenicity might facilitate survival of Y. pestis during the period of transmission from flea-to-host [38].
Although some details related to regulatory networks and the physiologic consequences are understood, additional details remain to be elucidated. For example, Y. pestis escapes from innate immunity defense by inhibiting cytokine production or the LPS-induced inflammatory response [52,53], but more comprehensive analysis of (epi)genomic comparison should be performed on Y. pestis strains with different host ranges to further establish the physiologic basis to this extraordinary aspect of the Y. pestis lifestyle. The interactions between chromosome and plasmids, especially the horizontally-acquired plasmids, between plasmids, or the contribution of small open reading frames (sORFs) [54], will strengthen our understanding of this phenomenon.
KNOWN 2: GENETIC DIVERSITY OF Y. PESTIS
Y. pestis was shown to be a genetically-monomorphic species with the advent of molecular genotyping [6,55]. By using multi-locus sequence typing (MLST) methods [56], which is frequently used in pathogen genotyping and spread analysis, it is apparent that within-species diversity cannot be determined, i.e., all strains of Y. pestis have an identical sequence type based on the sequences of six housekeeping genes [6].
To determine the genetic diversity of Y. pestis before the appearance of next-generation sequencing technology (NGS), researchers investigated multiple types of genomic variations, including different regions (DFRs), variable number of tandem repeats (VNTRs), and clustered regularly interspaced short palindromic repeats (CRISPRs), while developing corresponding genotyping methods. The DFR method was built on the presence and/or absence of 23 genome fragments and distinguishes 909 Y. pestis natural isolates into 32 genotypes [57]. According to Platonov [58], DFR-typing of 275 Y. pestis strains from the CIS Natural Foci identified 56 novel genomovars, indicating that the discriminatory power of this method is high enough to distinguish between subspecies, populations, and even strains circulating in natural plague foci. There are three CRISPR spacer arrays in the Y. pestis chromosome. According to the composition of spacers in these CRISPR loci, 125 Y. pestis representative isolates from China, the former Soviet Union, and Mongolia, were classified into 12 types [59]. Multiple loci VNTR analysis (MLVA) methods provide high discrimination power because integrated variation information of multiple VNTR loci have high mutation rates, and therefore could be used in outbreak investigations to provide clues on source tracing [60]. The rapidly-mutated VNTR loci are prone to parallel or reverse changes, which makes the deep branches of the phylogeny less robust, hence VNTR loci are not suitable for making inferences about the long-term evolutionary dynamics of the bacterial population.
Advances in NGS technology have reduced the cost and speed of bacterial whole genome sequencing, which facilitated deciphering hundreds of Y. pestis genomes of natural isolates and robust high-resolution genealogy was rebuilt based on genome-wide variations through population genetic methods. Results from whole-genome sequencing verified that Y. pestis is a young and monophyletic species that evolved from Y. pseudotuberculosis between 2600 and 28,000 years ago [61], with recent analysis narrowing it down to about 6,000 years ago [62]. The average pairwise genetic distance among any two natural isolates of Y. pestis is 126 single nucleotide polymorphisms (SNPs) [61]. It has been reported that Y. pestis has a decay genome that resulted from adapting to the nutrient-rich environment of the new survival niche (i.e., rodent host blood). The genome content composition supports the following hypothesis: compared with other species, Y. pestis has a relatively closed pan-genome with a length of 5.4 Mb, but a rapidly reduced core genome size of 3.5 Mb based on the genomes of 133 Y. pestis global isolates with average genome size of 4.6 Mb.
Currently, 33 phylogroups in 5 main branches of Y. pestis have been identified (Fig 1). Branch 0 is a root lineage of Y. pestis, which contains several ‘atypical’ groups. The 0.PE2 and 0.PE5 groups are sub-species of Y. pestis and termed ‘pestoides’ [63]. Strains of the 0.PE4 group are assigned to the biovar, Microtus, and are known as highly virulent in their main hosts (Microtus spp.) and laboratory mice, but virulence is attenuated in larger mammals, such as guinea pigs and humans. The 0.PE3 group contains only the Angola strain, which has the largest number of strain-specific SNPs (n=437) when compared with the average number of 126 SNPs across all species, although the reason for this finding is unknown [61,64]. The 0.PE7 group represents the oldest group among all modern Y. pestis natural isolates and includes two strains that were both isolated from Qing-Tibet plateau of China in the 1960s [61]. One 0.PE7 strain was isolated from a human plague case, suggesting that Y. pestis acquired pathogenicity to humans in the very early stages of Y. pestis evolution, which has subsequently been confirmed by aDNA sequencing analysis [9].
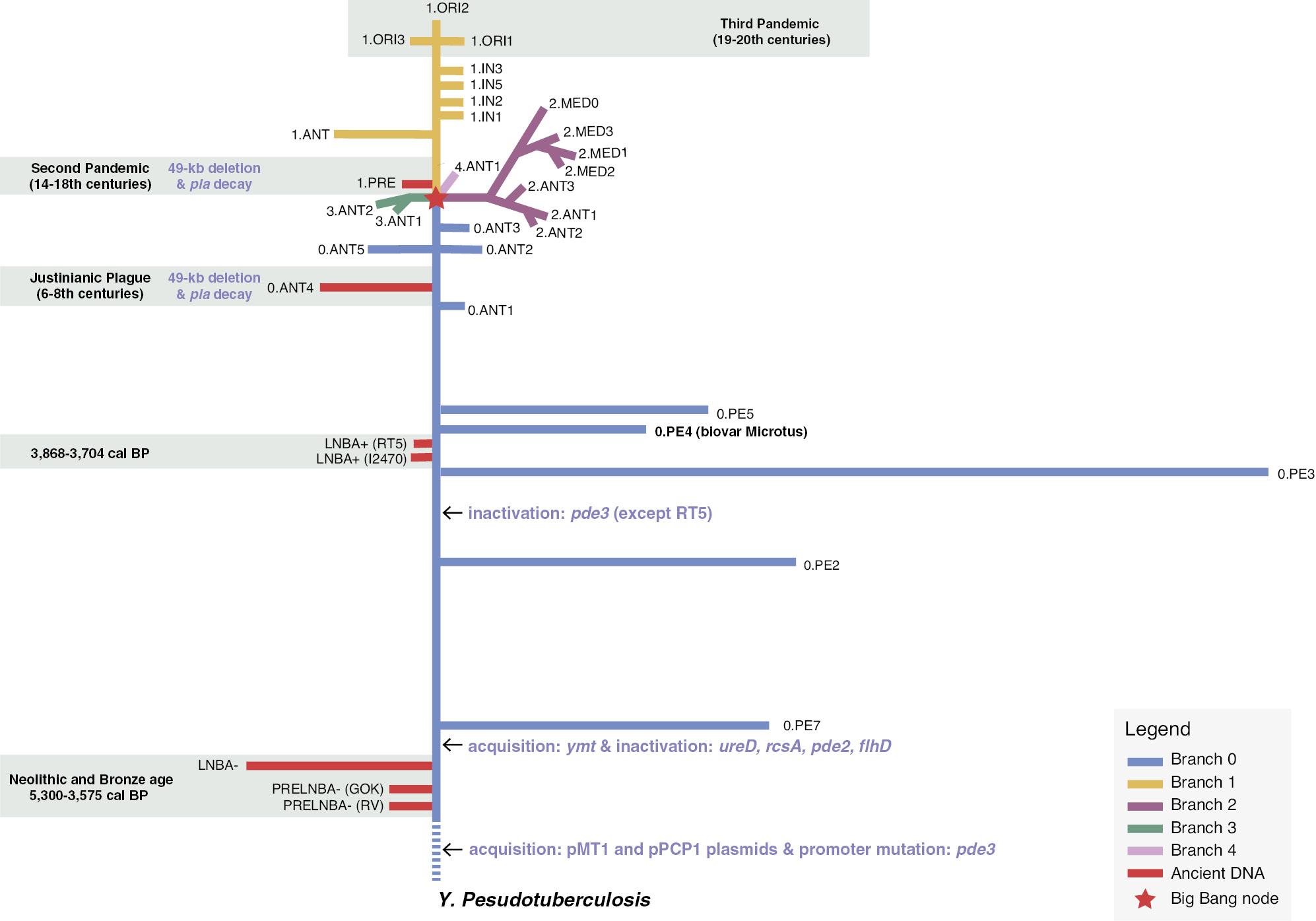
Schematic phylogenetic tree of Y. pestis with Y. pseudotuberculosis at the root representing the ancestor of Y. pestis. Five major branches (branches 0-4) are shown in different colors with ancient DNA-related lineages colored in red (see color legend at the bottom-right corner). The ‘Big Bang’ node giving rise to branches 1-4 is marked by a star. Shaded areas indicate phylogroups associated with prehistoric plague or three historically-recorded pandemics. The key events of plasmid acquisition and gene gain/inactivation/loss during evolution of Y. pestis are also displayed.
The Justinianic plague was the first plague pandemic to be described in historical records and was caused by an extinct lineage (0.ANT4; Fig 1) [65]. After the appearance of the other three 0.ANT lineages (0.ANT2, 5, and 3), a ‘Big Bang’ node of Y. pestis emerged in the early 14th century, from which branches 1-4 radiated [65,66]. Based on a synthesized analysis of archaeological, historical, and ancient genomic data, Spyrou et al. [67] found that ancient Y. pestis strains from the Kara-Djigach tombs (the Tian Shan region of North Kyrgyzstan) dated 1338-9 are situated exactly on the Big Band node [67]. After the ‘Big Bang’ node one SNP accumulated along branch 1 an ancient genome from Laishevo (Tatarstan) (LAI009) and one more SNP after LAI009 was observed from the remnants of the Black Death strain originating at the beginning of the 2nd pandemic [66,68,69], thus the Big Bang event occurred immediately preceding the Black Death. Because the Kara-Djigach genomes came from a region in which 0.ANT, including 0.ANT3, the closest old ‘cousin’ of the Big Bang are currently circulating, it is thought that the ‘Big Bang’ must have occurred in a marmot reservoir of the Tian Shan region [67]. Thus, the spatio-temporal origins of the Big Bang have been established. One of the newborn lineages from the Big Bang is branch 1, the strains of which are associated with the 2nd pandemic (commencing with the Black Death) and the 3rd pandemic (commencing in Yunnan in 1772 and becoming global in 1894). Branch 1 is currently the most widely-distributed lineage of Y. pestis that currently thrives in natural plague foci in Asia, Africa, and the Americas, and most likely thrived in Europe in the late medieval and possibly early modern periods [70,71]. Specifically, the 2nd pandemic waves were caused, with one exception, by the now defunct branch 1A and the sub-lineages of branch 1A, while the 3rd pandemic was spread by 1.ORI lineage strains (derived from branch 1B), which appears to have left Europe in the 1360s, across the globe through steamship transportation at the end of the 19th century [71,72]. There are 90 SNPs and approximately 380 years separating the Big Bang and the Great Plague of Marseille (1720-2), implying 1 mutation in 4.2 years within branch 1A. Branch 2 is split into 2.ANT and 2.MED lineages, with the former strains circulating in Nepal, China, and Mongolia, while 2.MED is found throughout Asia from Caucasus, Caspian, and the Volga-Ural region in the West via western Kazakhstan, Turkmenistan, and northern Kyrgyzstan into China and Mongolia. The reported number of branch 2 genomes is the second largest in Y. pestis, and only less than branch 1. Strains of branch 3 are only found in Gansu Province and Qinghai Province of China and Mongolia, with 12 genomes reported as of April 2022. Strains of branch 4 are only found in the Altai region of Russia and Mongolia, with 11 genomes reported as of April 2022 [61,73,74].
The genomes of ancient DNA (aDNA) provide genetic information for historically-extinct populations, and therefore are important in bacterial evolutionary research as a molecular fossil [75]. Following the successful sequencing of the first whole aDNA genome from the Black Death victims in London (1349) in 2011, there are more than 110 publicly-available ancient genomes of Y. pestis identified as of April 2022 and 7 extinct lineages (Fig 1) [62,66,67,76]. This finding extends our knowledge of human plague infection to the late Neolithic and Bronze ages and beyond to the early and later medieval periods across Eurasia and North Africa. Furthermore, studies of ancient genomes have yielded valuable information on the early adaptive evolution of this bacterium, such as the development of flea-borne transmission, and also revealed changes in genomic profiles during pandemics, such as convergent evolutionary signals, the 49-kb deletion, and pla decay at the end of the first two pandemics (Fig 1) [66,77].
UNKNOWN 2: EVOLUTIONARY DYNAMICS AND DRIVEN FORCE
Although the genealogy of Y. pestis has been well-defined based on population genomic studies, driving force shaping, such as genealogy and the detailed dynamics during its evolution, are vague. Whole genome-wide SNP analysis suggests that the evolutionary process of Y. pestis is generally neutral, i.e., most of the observed mutations accumulated randomly [61]. After considering multiple types of variation, such as indels and gene gain/loss, our current understanding might change, as was the case with the Salmonella enterica serovar, Paratyphi A. The population genetic analysis combining variations of SNPs, indels, and accessory genomes provided evidence of transient Darwinian selection during its evolution [78]. In addition, by using SNPs and indels, five mutation hotspots have been identified with strong selection signals in 78 Y. pestis isolates [79]. Mutations at one hotspot, the rpoZ gene, might affect the vector behaviors and have proved to be closely related to climate changes. In fact, 19 of all 130 mutations (14.6%) involved in mutation hotspots exhibited selection signals [79], suggesting that natural selection has a more important role than previously thought.
Therefore, reconstructing the evolutionary dynamics using combined variations, including SNPs, indels, and gene gain/loss, is still a challenge. Another challenge is the association between genetic variations and niche factors, including climate, soil components, hosts, and vectors, for inferring natural selection forces. By understanding how Y. pestis interacts with the environment we can begin to understand the natural survival strategies and learn how the genome is shaped, thus promoting the development of novel countermeasures for plague prevention and control.
The evolution of virulence in Y. pestis is also an important and unsolved issue. Y. pestis is a clone that derives from its ancestor species, Y. pseudotuberculosis, with few intermediate lineages between the two species. Therefore, it is difficult to infer the step-by-step evolutionary scenario. Based on comparative genomic analysis, we know that Y. pestis acquired two plasmids (pMT1 and pPCP1), including the subsequent gain of the Yersinia murine toxin (ymt) gene in the pMT1 plasmid, and inactivated numerous functional genes (ureD, rcsA, flhD, pde2, and pde3; Fig 1) [7,77,80,81]. The order in which these events occurred and the fitness advantages during the evolution are not clear. Additionally, there is no satisfactory explanation for the distinct animal virulence across different phylogroups of Y. pestis, except for a few clues that relate selection pressures with fine genomic changes, such as frameshift indels and copy number variations [82].
KNOWN 3: DISTRIBUTION AND ECOLOGY OF NATURAL PLAGUE FOCI
The existence of plague natural foci has long been recognized, as far back as 1910-1911 during the Manchurian plague epidemic, which resulted in 50,000-60,000 deaths. Zabolotny [83] reported that the epidemics were caused by a spillover of the Y. pestis population from the natural Marmota sibirica plague foci.
Natural foci consist of Y. pestis, hosts, vectors, and the local environment, and are based on the food chain and spatial interaction between Y. pestis, hosts, and vectors, which then form the biomes of plague in evolution and finally establish the biogeographic community suited to the specific local environment [84,85]. The global distribution of plague is extensive and has been observed on all continents, except Antarctica, with natural foci distributed widely in Asia, Africa, and the Americas (Fig 2) [86,87], and covering wet and dry regions, grasslands, deserts, plateaus, and plains.

In Asia, the active foci distributed in Central Asia (mainly in the Lake Balkhash region of Kazakhstan), Mongolia, China, Vietnam [86,88,89], and Africa, and the natural foci exist in broad areas (mainly in the southern and eastern regions) but is particularly prevalent in Madagascar, Tanzania, and the Democratic Republic of Congo [86]. In the Americas plague is endemic in Brazil, Bolivia, Ecuador, Peru, and the United States [86], and in Europe and Oceania there are no natural plague foci today, but most European countries were severely affected during the second and third pandemics [90].
Mammals are the most common host species with 351 species globally that act as hosts [87]. Among all these species, 279 rodent species have been identified as plague carriers. According to the strong adaptability to a wide variety of lifestyle, environments, and high reproductive capacity, rodents have a worldwide distribution (excluding Antarctica) and have an integral role in the Y. pestis circulation process [87,91]. Y. pestis circulating within natural foci is thought to be guaranteed with the existence of hosts through three processes: preservation; accumulation; and dissemination [87]. The primary rodent hosts that persist in the region are generally resistant to the bacteria and experience low level bacteremia when infected, which results in the pathogen preservation process. The accumulation process occurs when Y. pestis is transmitted to sensitive secondary and tertiary rodent hosts from primary rodent hosts via fleas and the secondary and tertiary rodent hosts generate a high level of bacteremia. Rodent overcrowding is a key contributing factor to dissemination of Y. pestis [92].
Due to geographic and climatic diversity, host distribution varies globally with approximately 70 species known as main reservoir species concentrated in the regions corresponding to current plague foci, such as western North America, eastern South America, eastern Africa, central Asia, and southeast Asia [87].
In the natural foci of plague, only fleas are the main vectors. Under natural conditions, the plague pathogen has been detected in 280 species and sub-species of fleas belonging to 62 genera [93].
Except for the indispensable roles of hosts, vectors, and Y. pestis for developing natural plague foci, environmental and ecologic parameters also play key roles in this process. At the end of the last century, it was proposed that global climate dynamics are responsible for outbreaks of infectious diseases, and others warn that long-term global warming increase the risk of acquiring such diseases. Parmenter et al. (1999) proposed the trophic cascade model in ecology and hypothesized that increased precipitation during the winter-spring period in arid and semi-arid regions increase ecosystem productivity, and further give rise to a greater population of mammal plague hosts and insect vectors [94]. Subsequent articles related to the trophic cascade model proved the validity of the hypothesis [95], and found additional complexity between the elements of the model, such as the lag effect of precipitation, bottom-up regulation, and immediate climate effects on plague prevalence [96,97].
Maintenance of plague foci depends on an entire suite of rodent hosts and their associated fleas [98], and climate affects the occurrence of plague by affecting rodent distribution, vector burden, population density, and ultimately susceptible population exposure to Y. pestis [99–101]. Unfavorable climatic conditions or extreme weather events due to climate change lead to reduced productivity and famine, thus leading to human migration followed by rodents in search of water and food, which elevates human-rodent interactions and finally increases the risk of human plague [102,103]. Additionally, climate influences replication cycles of pathogens or alters the mode of transmission, which in turn leads to changes in the prevalence of plague [104].
The patterns of climate change differ from local-to-regional levels and ultimately to the global level with humidity, rainfall, and temperature studied in detail at the local level. Studies investigating climate and plague epidemiology in India during 1898 and 1949 revealed that the timing of plague outbreaks was associated with seasonal changes in humidity [105], while work examining the climate drivers of plague in the West Nile region of Uganda concluded that the number of suspected cases in this region were negatively associated with dry season rainfall and positively with rainfall prior to the plague season. At high elevations, plague risk is positively associated with rainfall during the winter and spring, and negatively associated with rainfall throughout June [106,107]. Conversely, environmental predictors of plague in Vietnam showed that the risk of plague increases during the dry season when rainfall is low [108]. In addition, a study investigating the factors influencing the global transmission velocity of plague during the third pandemic concluded that temperature exhibits a non-linear, U-shaped association with spread speed [109].
There are also studies at the regional level. Research involving the human plague in the USA showed that the Pacific Decadal Oscillation, together with previous plague levels and above-normal temperatures, explains much of plague variability [110]. Research on the connection between the North Atlantic Oscillation (NAO) and plague in pre-industrial Europe showed that the pathway from climate change-to-plague incidence is distinctive in its spatial, temporal, and non-linear patterns. The NAO-plague correlation in the Atlantic-central Europe region primarily remained positive, while the correlation in Mediterranean Europe alternately switched between positive and negative [102].
UNKNOWN 3: ECOLOGIC INTERACTIONS OF Y. PESTIS, HOSTS, AND NATURAL ENVIRONMENTS FOR LONG-TERM SURVIVAL OF NATURAL PLAGUE FOCI
Because of the scarcity or complete absence of controlled studies before the turn of the last century, there are obstacles in seeking the associations between the global scale climate drivers and plague outbreaks. Research on the relationship between volcanism and global plague pandemics has suggested that all three pandemics coincided with periods of significant volcanic activity, and a series of connections between volcanism and plague are possible [111].
The ability of Y. pestis to colonize and propagate in the flea gut prior to transmission to a new host is well-established [112]. Within an inveterate rodent population, enzootic (maintenance) plague episodes ensure that Y. pestis is passed through a partially-resistant enzootic host population by fleas and upon transmission into epizootic (amplifying) hosts, plague rapidly spreads [113]. Plague is therefore circulating in associated hosts prior to re-emergence in the human population [114,115]. Although this rat/flea model has endured as the plague reservoir for transmission to humans, the rat/flea model is likely to constitute only one mode of transmission because the general picture has been oversimplified [116].
A remarkable aspect of epizootic plague biology is the fact that Y. pestis displays inter-epizootic cryptic periods of quiescence [117,118] between plague outbreaks in rodents or humans. In locations where there have been no human cases or mass rodent die-offs [114,119], plague often re-emerges decades later, such as occurred in Algeria, Libya, Madagascar, and India 57, 25, 60, and 30 years ago, respectively. In the Xinjiang province in China, the Junggar Basin plague focus was discovered in 2005 following continuous surveillance and this plague focus was in the cryptic quiescence period since 1949 [120].
Reports of long inter-epizootic periods during which time Y. pestis is absent from the host and vector populations have largely been overlooked, possibly because it has been suggested that once Y. pestis evolves to colonize insect and mammalian vectors, Y. pestis becomes host-dependent [121]. It is more likely that Y. pestis adopts a ‘sit and wait’ lifestyle [122] in the environment in which Y. pestis can survive under highly diverse ecologic conditions that compare favorably with the lifestyle of the near identical (∼98% DNA identity), free-living soilborne gastrointestinal pathogen ancestor, Yersinia pseudotuberculosis, from which Y. pestis evolved [6,61,65,123].
Where then does Y. pestis persist during these quiescent periods and which factor(s) result in epizootics? There are several hypotheses to explain this phenomenon, including spillover of plague from the primary hosts-to-highly susceptible rodents with a high mortality rate [124]. This hypothesis would exacerbate transmission from low level and undetectable epizootics within the main host by changes in ecologic and environmental conditions, thus resulting in unexpected fluctuations of host and vector abundance and/or behavior [113,124,125]. There may also be the formation of the non-culturable L-form of Y. pestis in soil or survival in protozoa or biofilm formation abiotically or biotically on the surface of nematodes [126–129]. A number of microorganisms are resistant to soil-dwelling free-living amoebae predation [130]; this is also true for Y. pestis [131]. Markman et al. [132] and Benavides-Montañ
Another facet of interaction between Y. pestis, hosts, vectors, and environmental and ecologic parameters is which natural drivers select the variants of the bacterium and how the gene mutations in these variants impact the persistence of plague foci. For example, colder and drier winters may select for rpoZ mutants, which rapidly form biofilms and subsequently promote rapid transmission by infected fleas [79]. Corollary studies are warranted to elucidate the mechanisms underlying natural plague foci.
KNOWNS 4: TRANSMISSION OF Y. PESTIS BY FLEAS
Bubonic plague, as a vector-borne disease, and the key role of the flea in the transmission of Y. pestis were recognized soon after the discovery of Y. pestis. Several possible transmission mechanisms were considered, initially using a model in which groups of fleas were allowed to feed on a rodent dying of septicemic plague, then transferred to naïve rodents [137]. Transmission results if the fleas are transferred within a few days after being infected during a blood meal, but transmissibility wanes rapidly beyond a few days. This process is now referred to as early-phase transmission. A few years later, a second mode of transmission was discovered, the well-known ‘blocked flea’ transmission model [112].
Both modes of transmission stem from the marked propensity of Y. pestis to rapidly auto-aggregate in the flea digestive tract. Large masses of bacteria can form in the midgut and localize to the proventricular valve in the flea foregut within a few hours after an infectious blood meal [138,139]. Initially, the bacteria appear to be associated with a semi-fluid, serous matrix, and in some fleas an aggregate can extensively occupy the lumen of the proventriculus within a few days. Although long assumed to be mechanical (transmission via contaminated mouthparts), the infected foregut is the more likely source of bacteria transmitted during the early phase. According to this model, a heavy proventricular infection is sufficient to transiently impede blood flow, resulting in some backflow of contaminated blood into the bite site [138,140]. Because the early proventricular aggregates are relatively soft and fragile, however, the early proventricular aggregates are readily dislodged and washed back into the midgut by the incoming blood pressure, thus terminating early-phase transmission. The second mode of transmission, the well-described blocked flea mechanism, is phenomenologically the same (regurgitation from the foregut) except in this case the proventricular aggregate is more cohesive and firmly entrenched in the proventriculus because the proventriculus severely impedes the passage of blood into the midgut.
Fundamentally, the flea-borne transmission phases reflect a continuum of Y. pestis biofilm development in the proventriculus. Maturation of the biofilm over the following days notably involves production of a polysaccharide extracellular matrix that acts to stabilize Y. pestis aggregates, making the aggregates more cohesive and firmly fixed to the proventriculus [141]. A transmission-competent, partially-blocked stage develops first [142]. At the end stage of complete blockage, proventricular biofilm prevents feeding, but continuous probing and feeding attempts by a blocked flea result in more efficient regurgitative transmission.
Biofilm development is a common strategy of many bacteria to adapt to nutrient-limited moist environments and is characterized by the formation of surface-adherent dense microcolonies that are embedded within an extracellular polymeric matrix. Metabolic adaptation of Y. pestis to the protein- and lipid-rich, but carbohydrate-poor flea gut induces biofilm formation. The regulatory and development pathways underlying this process are complex, as reviewed recently [143], but culminate in upregulation of the Hms genes, which act to produce the aforementioned polysaccharide component of the biofilm matrix [138,144]. Y. pestis mutants lacking the hmsHFRS operon are unable to block fleas and are unable to synthesize the polysaccharide extracellular matrix stabilizing Y. pestis aggregates in the proventriculus [139,145]; however, a hmsR mutant is transmissible by the early-phase mechanism [146,147].
Differential biochemical characteristics of vertebrate blood and the speed at which vertebrate blood is digested strongly affects the ability of Y. pestis to colonize the flea gut and be transmitted [140,148]. For example, rodent fleas infected using rat blood are much better early-phase transmitters than fleas infected using mouse blood [140]. Furthermore, an augmenting effect of Hms-dependent biofilm formation on transmission (corresponding to evidence of partial or complete blockage) can be seen as early as 3 days after an infectious rat blood meal, suggesting that early-phase and proventricular blockage-dependent transmission overlap temporally. The initial bacterial aggregates following an infected rat blood meal are associated with a viscous mixture of undigested hemoglobin and red blood cell components that results in a much more tenacious proventricular colonization as well as reflux of the infected mixture into the esophagus [140]. This condition enhances transmission in the early phase.
Reliance on flea vectors for transmission between mammalian hosts is an evolutionary recent adaptation, arising within the last 3000-6000 years since Y. pestis emerged from its closely related progenitor, Y. pseudotuberculosis. This abrupt switch to arthropod-borne transmission was possible because transmission required just a few, discrete genetic changes [77]. Pseudogenization and functional loss of three genes extended the pre-existing biofilm capability of the Y. pseudotuberculosis progenitor to the flea gut environment because the loss results in increased intracellular levels of cyclic-di-GMP, a universal bacterial inducer of biofilm development. Two other genetic changes were also important. One genetic change was an additional gene loss that eliminated enzyme activity (urease) toxic to fleas [22], but not related to flea blockage. The other genetic change was the acquisition of a new gene present on the Y. pestis-specific plasmid encoding a phospholipase D enzyme (Ymt) that protects Gram-negative bacteria from a toxic product produced by the flea [11,149]. Making these same five changes in a modern Y. pseudotuberculosis strain was sufficient to enable the strain to block fleas [77].
The evolutionary road to flea-borne transmission was a stepwise process. The earliest Y. pestis strains, characterized by genome sequencing from infected human skeletal remains dating from the Stone Age, already had most, if not all the plague-related virulence factors. Most of the Stone Age strains had only one of the three biofilm-enhancing genetic changes still had urease and had not acquired Ymt [80]. This finding led to the conclusion that these strains could be transmitted by fleas, but to an extent that was not compatible to sustain a vector-borne cycle for a long period of time, at least in relation to blockage. It is now known that the importance of Ymt for bacterial survival in the flea gut is host blood-dependent. Ymt-negative Y. pestis survives well in fleas infected using blood of the brown rat (Rattus norvegicus) [149]. This new finding, together with the increased early-phase transmission efficiency associated with brown rat blood infections and the biofilm-enhancing effect of the one genetic change, suggest that a rudimentary rat flea-brown rat transmission cycle is possible. Interestingly, the northern China habitat of R. norvegicus overlaps geographically with the emergence of Y. pestis. Acquisition of Ymt and accumulation of the remaining gene losses described above occurred by the Bronze Age, fully enabling flea-borne transmission and significantly extending the host range [9,149].
UNKNOWNS 4: COMPLEX INTERACTION BETWEEN YERSINIA PESTIS AND FLEAS
While much has been learned, many gaps remain in our understanding of Y. pestis transmission. The general transcriptional responses of Y. pestis in adapting to the flea gut and the flea to infection have been characterized [38,150], but specific gene induction steps and molecular mechanisms required to produce a transmissible infection have yet to be fully elucidated. Key factors at the flea-Y. pestis-mammalian dermal interface that determine successful transmission have not been well-characterized. Vector competence differences between flea species and how this relates to enzootic and epizootic plague are not fully understood. Thus, questions ranging from individual Y. pestis-flea interactions to the complex ecology of plague involving many flea vectors and hosts are still ripe fields for further research.
A prime topic is biofilm formation in the flea. What is the molecular and biophysical basis for the rapid aggregation of Y. pestis upon entering the flea gut and what is the make-up of the extracellular matrix that surrounds these aggregates? These features of a vector-specific life stage may protect Y. pestis from antibacterial factors in the lumen of the flea digestive tract, which is a hostile environment for Gram-negative bacteria. Furthermore, Y. pestis is transmitted in association with the extracellular matrix. There are clear differences in the pathogenesis following flea-bite transmission versus intradermal injection of in vitro culture-grown Y. pestis. Specifically, a chronic intradermal infection often follows transmission by fleas but does not occur following intradermal injection [151]. Differences in antigenic makeup between flea- and culture-derived bacteria, as well as the presence of extracellular matrix may be responsible. Flea saliva is also injected into the bite site but the effect on nascent infection and immunity have not been thoroughly investigated. Mice and rats mount an immune response to flea salivary components, but the immune response does not appear to significantly affect productive transmission [152].
How the biochemical characteristics of host blood can so strongly influence infectivity and transmission is another unknown. The rate at which a particular type of blood is digested by the flea and the concomitant generation of antibacterial digestion products appear to be important, but the molecular mechanisms are yet to be identified. For example, the importance of the protective effect of Ymt varies with the blood meal source [149], but the identity of the relevant target(s) of this phospholipase D in the flea gut remains elusive.
Y. pestis is a generalist, able to produce a transmissible infection in many different flea species by the same mechanism; however, fleas vary widely in vector competence and transmission efficiency by both early-phase and blockage mechanisms [153]. The reasons for this finding are not clear, although differences in digestion kinetics and processing of host blood, feeding frequency, excretion rates, and foregut anatomy are likely influencing factors [138]. The incidence and rate of complete blockage development are much lower for some fleas than others, leading to proposals that early-phase transmission is more important from an ecologic perspective in some flea-rodent transmission cycles. Blockage rate comparisons between different fleas have been based on a limited number of studies with small sample sizes and variable experimental conditions, making conclusions tentative and sometimes discordant or misleading [154].
Quantitative evaluation of the relative efficiency of the early-phase and blockage-dependent transmission modes for different fleas merits systematic re-examination using standardized protocols with appropriate controls [137,153]. The infectious blood source should also be considered. For example, recent early-phase transmission comparisons have all been based on fleas infected using brown rat blood. As described above, this blood source induces an initial severe foregut infection highly favorable for early-phase transmission, which does not occur with other host bloods. Ideally, future studies would start with a single cohort of fleas infected using the usual blood source, which would then be monitored for early-phase transmission, blockage development, and blockage-dependent transmission at different times after infection. These studies would provide more reliable vector competence data for mathematical models of enzootic and epizootic plague.
KNOWN 5: VIRULENCE FACTORS AND ROLES IN THE PATHOGENESIS OF Y. PESTIS
Three pathogenic Yersiniae share several virulence mechanisms, among which the T3SS, encoded by a 70-kb plasmid (pCD1 in Y. pestis and pYV in Y. enterocolitica and Y. pseudotuberculosis) is essential for full virulence in all three pathogens. Although closely related in terms of evolution, these pathogens are extremely diversified in clinical symptoms, ecologic niches, and typical infection routes. Y. pestis is an etiologic agent of plague that has caused greater than 200 million deaths in the past 3 pandemics, whereas the other 2 cause only self-limited gastrointestinal diseases. In addition to acquisition of two virulence-associated plasmids (pMT1 and pPCP1), massive gene losses played more important roles than gene acquisitions in the virulence evolution of Y. pestis from the acknowledged ancestor Y. pseudotuberculosis [155].
Type III secretion system (T3SS)
Gram-negative bacterial T3SS assembles a macromolecular device (injectisome) that delivers virulence effectors into eukaryotic cells. Yersinia T3SS is temperature- and contact-dependent, and the secretion of effectors are triggered at mammalian temperatures only when contact with host cells occurs. Y. pestis virulence factors exhibit various enzymatic activities that disrupt the eukaryotic cytoskeleton and host immune signaling to promote bacterial survival and replication.
LcrV is the component of the T3SS injectisome needle tip structure and LcrV also induces production of suppressive interleukin (IL)-10 [156]. YopH is a potent tyrosine phosphatase that dephosphorylates a variety of functionally-distinct substrates. YopH inhibits T cell and B-lymphocyte activation via dephosphorylation of Lck and ZAP70, the major signal transducer for the T cell antigen receptor (TCR), enabling blockage of the first step in TCR binding and suppressing the immune response against Yersinia [157]. YopE, YopT, and YpkA, all of which belong to a large family of bacterial toxins that target the Rho family of small GTP-binding proteins (Rho GTPases), inhibit phagocytosis by professional phagocytes. YopE inactivates multiple Rho GTPases, including RhoA, Rac1, and Cdc42 [158,159]. YpkA is inactive in bacteria and is activated by binding to the co-activator, actin, in eukaryotic cells [160]. Upon activation, YpkA undergoes autophosphorylation and phosphorylates Gαq (the α subunit of heterotrimeric G proteins) [161], vasodilator-stimulated phosphoprotein (VASP) [162], and otubain-1 to disrupt actin cytoskeleton. YopT acts as a papain-like cysteine protease that removes the prenyl group from RhoA, RhoG, Rac1, and Cdc42, and releases these GTPases from the membrane that lead to inactivation [163]. YopT has been shown to contribute to the anti-phagocytic activity of bacteria but is not essential for the virulence of pathogenic Yersinia. This finding may be due to the fact that YopT is functionally redundant in the presence of YopE and YpkA [158].
Recognition of Y. pestis by host nod-like receptors (NLRs), including NLRC4, NLRP3, and pyrin, induces the formation of inflammasomes, leading to caspase-1 activation, IL-1β processing, and secretion and cell death. YopM enables bacteria to avoid this host innate immunity strategy by directly binding caspase-1 to inhibit caspase-1 activation and inflammasome maturation [164]. Furthermore, YopM interacts with pyrin and kinases, RSK1 and PKN1, the negative regulators of pyrin, to inhibit the activation of the pyrin inflammasome that is triggered by the RhoA-inactivating enzymatic activities of YopE and YopT [165,166]. YopM has a nuclear localization signal at the carboxyl terminus [167] and Y. pestis infections cause a systemic depletion of natural killer cells in mice in a YopM-dependent manner [168].
YopJ functions as an acetyltransferase that inactivates nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) pathways by acetylating MAPK kinase [169]. Once translocated into host cells, Yop effectors act in a finely-tuned and coordinated manner to hijack various host signaling pathways to thwart the innate immune response [158]. It has also been reported that YopJ has deubiquitination activity towards molecules in the NF-κB signaling pathway, including TRAF2, TRAF6, and IκB, to inhibit the inflammatory response [170]. Mutations of yopJ in Y. pestis show no obvious virulence attenuation, although greater virulence attenuation has been observed in enteropathogenic Yersinia [171–173].
YopK has an important regulatory role in Yop translocation and controls the rate and fidelity of Yop injection into host cells; a yopk mutant exhibits the Yops-translocation phenotype [174,175]. This regulatory mechanism is partially exerted by interaction of YopK with the YopB-YopD translocon, which prevents recognition of the Yersinia T3SS and inhibits NLRP3 and NLRC4 inflammasome activation [33]. YopK inhibits the attachment of Y. pestis to the host cells by binding to the extracellular matrix protein, MATN2, and disruption of the YopK-MATN2 interaction results in a Yops hyper-translocation phenotype similar to the yopK mutant [176]. YopK also interacts with the receptor for activated C kinase (RACK1) and this interaction promotes the phagocytosis resistance of Y. pseudotuberculosis [177].
Lipopolysaccharide and Pla
Y. pestis produces a short-chain, rough lipopolysaccharide (LPS) that lacks the O antigen due to mutations of genes involved in LPS biosynthesis. The O antigen confers resistance to complement-mediated phagocyte bacterial killing and is important for virulence of Y. enterocolitica and Y. pseudotuberculosis. Pla is a pPCP1-encoded protein protease that cleaves plasminogen to plasmin, which further degrades extracellular matrix proteins to promote bacterial dissemination. The proteolytic activities of Pla require rough LPS, but are inhibited by the O antigen, highlighting the selective advantage of rough LPS for Y. pestis [178]. Pla is also an adhesin that is specific for laminin and heparan sulfate proteoglycan and cleaves the C3 component [15,179]. Pla is not required for serum resistance. As the temperature shifts from 26°C to 37°C, lipid A of Y. pestis switches from a hexa-acylated to tetra-acylated form due to the loss of lpxL, which is responsible for addition of the secondary acyl chains to the tetra-acylated lipid A precursor [53]. Tetra-acylated lipid A is poorly recognized by Toll-like receptor 4 (TLR4), which results in lower toxicity compared to other Enterobacteriaceae grown at 37°C [52] and facilitates pathogen escape from host immune surveillance [180].
F1 Capsule
The F1 capsule is composed of a linearly-polymerized Caf1 subunit that is encoded by the pMT1 plasmid. F1 appears to have some role in anti-phagocytic activity, although the primary Y. pestis virulence factor that blocks uptake by phagocytosis is T3SS [181], and strains with spontaneous mutations or deletion of caf1 are still virulent [182,183]. The F1 antigen contributes to virulence in naturally-acquired bubonic plague but is not essential for pneumonic plague in a mouse or guinea pig infection model [13,181,184–187]. An anti-F1 antibody provides high protection against F1-positive, but not F1-negative strains [188].
Adhesion and invasion
Y. pestis is lymphophilic and prefers to attack some types of cells, such as innate immune cells, including macrophages, neutrophils, and dendritic cells (DCs) [189], as well as lung alveolar epithelial cells. The pH6 antigen (PsaA) adhesin contributes to the cell-type preference of Y. pestis. Two major receptors for Psa have been shown to be β1-linked galactosyl residues in glycosphingolipids and phosphocholine, and phosphatidylcholine in phospholipids, which are present on alveolar epithelial cells [190,191]; however, an in vitro cell infection assay showed that PsaA promotes resistance to phagocytosis rather than adhesion to mouse macrophages [192]. PsaA facilitates delivery of Yops via T3SS by mediating intimate contacts between host cells and bacteria, as do other adhesions, such as the attachment invasion locus (Ail) and Pla [193]. Mutation of the psaA locus results in a 100-fold increase in LD50 via intravenous challenge, but no attenuation by the subcutaneous route of infection [194,195].
Ail is a chromosomally-encoded small-membrane protein common to all three pathogenic Yersinia. The major adhesins (YadA and Inv) in Y. pseudotuberculosis are inactivated in Y. pestis, thus Ail is the primary adhesin of Y. pestis. An ail-negative mutant of the KIM5 (pgm -) was attenuated over 103-fold in intravenously challenged mice and deletion of ail in CO92 resulted in an approximately 105-fold attenuation in a rat model of bubonic and pneumonic plague [196–198]. Ail binds to the ECM components, fibronectin, laminin, and heparin [199,200], and is critical for invasion and Yop delivery into host cells. Ail also binds to the complement inhibitor, C4b-binding protein, and vitronectin, a host protein that is critical in cell attachment, fibrinolysis, and inhibition of the complement system [198,201,202]. Y. pestis Ail shows higher binding affinity to ECM substrates than Y. pseudotuberculosis Ail because of two amino acid differences, and the rough LPS of Y. pestis increases accessibility of Ail to eukaryotic cells.
Yersiniabactin
Iron utilization by Y. pestis predominately relies on yersiniabactin (Ybt), which is encoded in a high-pathogenicity island within a 102-kb pigmentation (pgm) chromosomal locus common to the pathogenic Yersinia species. The ybt locus includes genes for Ybt siderophore synthesis (high-molecular-weight protein 1 [HMWP1], HMWP2, YbtD, YbtE, YbtS, YbtT, and YbtU) and Ybt uptake (YbtQ, YbtP, and Psn). Ybt has high affinity for ferric iron and is necessary for acquiring iron from transferrin and lactoferrin by Yersiniae [203]. The pgm - mutant or specific Ybt synthesis or uptake mutants are avirulent in subcutaneously-challenged mice, but are fully virulent in intravenously-challenged mice [45,204–206]. In the natural context of infection (i.e., a fleabite), a Ybt-negative mutant is also attenuated in virulence, but remains capable of causing fatal disease in some mice [45].
UNKNOWN 5: EARLY INTERACTIONS BETWEEN Y. PESTIS AND THE HOST CELLS: STILL LARGELY A MYSTERY
Y. pestis is a facultative intracellular pathogen that predominantly lives an extracellular life. In a typical fleabite transmission, Y. pestis bacilli are readily engulfed by professional phagocytes when initially entering the host. Y. pestis bacilli taken up by polymorphonuclear neutrophils (PMNs) can temporally survive and replicate within 3 days post-infection (dpi); however, the living Y. pestis bacilli can only be found in macrophages (Mφs) at 5 dpi [207]. The released bacteria from the disrupted host cells embark on an extracellular life thereafter. Thus, rapid adaption to the adverse mammalian host environment is critical for Y. pestis vis-a-vis arming itself with a variety of virulence factors, including the F1 capsule and the T3SS, to avoid phagocytosis and to paralyze the host immune response. An impressive feature of plague infection is that Y. pestis replicates without evoking a significant host innate immune response during the initial period of infection, termed the pre-inflammatory phase. Studies of bubonic plague rodent models revealed that Y. pestis replicates rapidly in draining lymph nodes (dLNs) near the infection sites with no detectable inflammation at the early stage (6–36 h post-infection [hpi]) of infection, then escapes from the bubo and is disseminated via the lymphatic circulation to establish systemic infections [50,208,209]. Pathologic studies on mouse primary pneumonic plague confirmed the biphasic feature, in which the infection begins with an immunosuppressive state in the first 24–36 hpi, then rapidly progresses to a highly proinflammatory state by 48 hpi [210].
Although the rapid adaption to adverse mammalian host environments is critical for the pathogenesis of Y. pestis, reports on the early interactions between the bacteria and the host immune cells are limited and remain to be clarified. It has been shown that Y. pestis preferentially targets host immune cells during infections. DCs, Mφs, and neutrophils are the most frequently infected cells, while T and B lymphocyte are less often infected [189], even though Yop effectors are delivered to nearly all types of cells in tissue culture [189,211]. In an intradermal (i.d.) infection of Y. pestis, a larger number of PMNs were shown to be recruited by 4 hpi and the majority of cell-associated bacteria were associated with PMNs, but very few bacteria interacted with DCs [212]. Neutrophil depletion and CCR7 knockout mouse experiments indicated that dissemination of Y. pestis from the dermis to the dLNs may not rely on DCs and PMNs. Another study showed that intracellular Y. pestis bacteria survive and replicate in cultured human PMNs, and a high percentage of the infected PMNs undergo apoptosis within 12 hpi. The PMNs containing Y. pestis can be recognized and internalized by autologous Mφs, in which Y. pestis survives and replicates following efferocytosis [37].
Recently, to describe the critical events occurring during the initial interactions between Y. pestis and the host innate immune cells, single-cell RNA sequencing (scRNA-seq) was utilized to characterize the composition and alterations of immune cells in the functional dynamics of dLNs during the early stages of bubonic plague, which revealed a dynamic immune landscape at single-cell resolution of murine dLNs in response to Y. pestis infection [27]. The data suggested that DCs respond to the presence of Y. pestis within 2 hpi, followed by the activation of Mφs and monocytes (Mons) and recruitment of PMNs to dLNs at 24 hpi. PMNs could be recruited to lymph nodes by CCL9 secreted by Mφs and Mons through CCR1–CCL9 interactions. When mice were treated with BX471, a CCR1 antagonist, prior to infection the number of PMNs in dLNs at 24 hpi decreased significantly in comparison to the untreated mice, suggesting that PMN recruitment by Mφs and Mons through CCR1 is important for host defense against plague. It is critically important to confirm these findings based on a scRNA-seq study in an animal bubonic model in future investigations.
Y. pestis biovar Microtus strains are highly virulent to mice, but avirulent to humans and other larger mammals. The underlying mechanisms responsible for this unique host-specific pathogenesis are far from clear and difficult to investigate due to biosafety and ethics issues. In vitro infection studies showed that human Mφs clear intracellular Y. pestis Microtus 201 more efficiently than murine Mφ RAW264.7 cells [213]. Given that survival in Mφs at the early stage is critical for the later establishment of Y. pestis infection, it is possible that the key events occurring during this period could lead to the host-specific pathogenesis of biovar Microtus strains. Higher bactericidal activity of human Mφs to Y. pestis 201 could partially explain the virulence attenuation of biovar Microtus strains in humans. Biovar Microtus strains of Y. pestis are more closely related to the progenitor, Y. pseudotuberculosis, than the strains of other biovars based on phylogenetic studies [72]. The products of the biovar-specific genome content could be involved in the direct bacteria-host interactions or control of the adaption to the hostile environments inside the host to contribute to the variation in virulence among different mammalian hosts.
KNOWN 6: DIAGNOSIS AND TREATMENT OF PLAGUE
The diagnosis of plague depends on epidemiologic information, clinical manifestations, and physical and laboratory examinations. Plague patients usually have contact history with infected animals or patients. In the clinical setting, Y. pestis infections usually present as bubonic plague with regional lymphadenopathy, primary pneumonic plague following direct inhalation of infectious respiratory droplets or aerosolized Y. pestis, and primary septicemic plague resulting from cutaneous exposure [214]. Some other less common forms of plague include local cutaneous ulcers at the entry site of Y. pestis in humans, plague pharyngitis, plague endophthalmitis, meningeal plague, secondary septicemia plague, and secondary pneumonic plague. Isolation and identification of Y. pestis from clinical specimens are critical for the accurate diagnosis in the laboratory (https://www.cdc.gov/plague/healthcare/clinicians.html) [215]. F1 capsular antigen and the corresponding antibody are usually targets for immunologic detection methods [216], including the F1 antigen hemagglutination inhibition test, passive hemagglutination test, enzyme-linked immunosorbent assays, and direct fluorescent antibody testing. A polymerase chain reaction (PCR) is often used for directly detecting Y. pestis in clinical specimens. Some point-of-care testing, such as immunochromatographic assays (ICAs), have been developed for on-site detection of Y. pestis or Y. pestis antibody.
The WHO Plague Manual: Epidemiology, Distribution, Surveillance and Control [214] and WHO guidelines for plague management [217] recommend a treatment scheme for the different forms of plague.
The U.S. Centers for Disease Control and Prevention recommend a new guideline for antimicrobial treatment and prophylaxis of plague [218]. This guideline gives detailed treatment schemes for different types of plague. To treat pneumonic plague [218], the first-line antibiotics for adults include fluoroquinolone (ciprofloxacin, levofloxacin, and moxifloxacin) and an aminoglycoside (gentamicin and streptomycin), while the alternatives are tetracycline (doxycycline), amphenicol (chloramphenicol), some fluoroquinolones (ofloxacin and gemifloxacin), aminoglycosides (amikacin, tobramycin, and plazomicin), and a sulfonamide (trimethoprim-sulfamethoxazole). For children aged ≥1 month to ≤17 years, the recommended first-line antibiotics are fluoroquinolones (ciprofloxacin and levofloxacin) and aminoglycosides (gentamicin and streptomycin). Alternatives are tetracycline (doxycycline), chloramphenicol, fluoroquinolones (moxifloxacin and ofloxacin), aminoglycosides (amikacin and tobramycin), and sulfonamides (trimethoprim-sulfamethoxazole). Further recommendations for treatment of bubonic plague and other less common forms of plague in adults and children, including prophylaxis for pre- and post-exposed adults and children, have also been described [218].
Y. pestis isolates with multiple drug resistance by a transferable plasmid have previously been reported in Madagascar [219,220]. Y. pestis is also considered to be a category A agent with bioengineered multi-drug resistant-Y. pestis with the potential to be used as a bioterrorism weapon [221,222]. There is therefore a need to develop precision treatment schemes using antibiotic combinations or other biotherapeutics, including monoclonal antibodies [223]. A potent inhibitor targeting LpxC [uridine diphosphate-3-O-(R-3-hydroxymyristoyl)-N-acetyl-D-glucosamine deacetylase], was reported to be potentially effective for treating plague [224–227]. Other potential drugs, including cationic antimicrobial peptides, anti-virulence drugs, predatory bacteria, phages, phage endolysins, and immunotherapies, are also under study [226].
UNKNOWN 6: ON-SITE DIAGNOSTIC TECHNIQUES: PRECISION TREATMENT USING DIFFERENT KINDS OF ANTIBIOTICS OR OTHER BIOTHERAPEUTICS
Although we have comprehensive immunologic and nucleic acid-based techniques for plague diagnosis in the laboratory, we still lack rapid, sensitive, and specific point-of-care testing for on-site detection and ultrasensitive assays in the laboratory for detecting traces of Y. pestis. Single molecule detection methods are being developed [228,229]. A microsphere labeled with carbon dots (CDs) and a colloidal gold-based lateral flow immunochromatographic assay combined with CRISPR/Cas-based nucleic detection for highly specific and sensitive detection of bacterial pathogens has been developed [230,231]. These highly-sensitive techniques are also easily developed as a point-of-care testing for onsite screening of potential pathogens.
KNOWN 7: LIVE ATTENUATED VACCINES AND SUBUNIT VACCINES
Attempts to create effective plague vaccines began soon after the microorganism was first identified. Most approaches involve inactivation of bacterial culture of wild-type isolates and the result was a commercial preparation of USP vaccine composed of formaldehyde-killed Y. pestis 195/P that has been used in the West for several decades. The major contributor to immunity elicited by this vaccine was likely provided by significant expression of the capsular antigen F1; however, this vaccine was reactogenic with short-term immunity. Importantly, the vaccine did not convey robust defense against pulmonary exposure to Y. pestis. Currently, the vaccine is not available [232].
The live plague vaccine (LPV) introduced by Girard and Robic via selection of attenuated variant EV76 was a breakthrough event, and saved numerous lives in Madagascar when first tested during the 1930 plague epidemic. Derivatives of this vaccine were widely used as the LPV prototype in many countries. This vaccine was never approved in the West due to safety concerns, but was widely used in the USSR [233] where it was administered to millions of individuals without any serious complications. LPV is currently in use in several counties of the former Soviet Union to immunize plague workers and those living in plague-endemic territories when plague is active [234]. The LPV in lyophilized form is easy to produce, store, and utilize; however, the major disadvantage is short-term protection that requires an annual booster. The reason for this phenomenon is not fully understood because live vaccines often elicit prolonged or even life-time immunity. Unraveling the mechanism underlying this LPV deficiency in human vaccination will be crucial for the development of an improved version of LPV, and likely for the development of a novel subunit plague vaccine [234]. Of note, a study involving humans vaccinated with EV NIIEG (EV76 version) using a panel of highly-pure recombinant Y. pestis antigens showed that both humoral and cellular immune responses to LcrV are generally poor. This finding was true even for donors who received multiple annual boosters over a long period of time [235]. Because LcrV is a major protective antigen of Y. pestis (section 5.1), it is thought that modification of LPV to elicit an enhanced response to LcrV may increase the level and length of protection.
Since the first publication demonstrating that recombinant LcrV provides protection in mice against infection with Y. pestis [236], several hundred articles have been published that described successful use of this antigen alone or in combination with F1 in development of plague subunit vaccines (PSVs) [237]. The LcrV provided most of the protection, while the addition of F1 further increased the level of immunity. The LcrV/F1 vaccines have been proven to work well in a number of formulations, such as purified antigens with different adjuvants, micro- and nano-particles, viral and bacterial vectors, and plants [237]. The most common animal models to test the protective properties of this specific PSV against bubonic and pneumonic plague include mice, rats, guinea pigs, and non-human primates. This subunit vaccine still needs optimization for human use, mostly with respect to selecting the most efficient adjuvant and/or method of antigen expression. The different versions of LcrV/F1 subunit vaccines have undergone or are in the process of phase 1 and 2 clinical trials; however, no phase 3 clinical trials have been conducted on the LcrV/F1 subunit vaccines thus far (https://clinicaltrials.gov/ct2/show/NCT00246467; https://clinicaltrials.gov/ct2/show/NCT00332956; https://clinicaltrials.gov/ct2/show/NCT01381744; https://clinicaltrials.gov/ct2/show/NCT05330624).
UNKNOWN 7: EFFECTIVE VACCINE FOR LONG-TERM PROTECTION: IMMUNE CORRELATES
The EV76-based LPV is attenuated mostly due to the lack of the pigmentation locus (Pgm), which contains a high-pathogenicity island. Therefore, some efforts have focused on improvement of such vaccines by introducing additional mutations. For example, the lpxM (msbB) mutant of EV NIIEG affecting the lipid A structure is less reactogenic for animals and provides prolonged multiplication in lymphoid tissue. These characteristics potentially improve both protective properties and immune duration of the existing LPV [238]. The usefulness of this approach should be verified in clinical trials. Mutations in many other genes have also been evaluated in an attempt to rationally attenuate Y. pestis for vaccine use, both alone and in combination with Pgm-negative variants [233,239]. The most promising variant was a triple-deletion mutant of Y. pestis CO92 impaired in lpp, msbB, and ail [240]. This mutant not only elicited strong humoral and cellular immune responses and robust protection in different animal models, but was safe enough for the Centers for Disease Control and Prevention to remove the mutant from the select agent list.
Overall, the central advantage of live vaccines compared to other vaccine types is the ability to elicit strong immune responses to several antigens, including antigens expressed in vivo, mimicking to some extent the initial course of natural infection. In addition, live vaccines are known to stimulate robust T cell immunity that contributes to protection against plague [241]. While the capsular antigen, F1, is a well-established protective antigen, the response to this polymeric abundant protein is overwhelming, and might be detrimental to the overall immunity. To increase the contribution of other antigens in the LPV, it might be beneficial to reduce the expression of F1 to decrease immunodominance.
Currently, plague subunit vaccines in clinical trials consist of the F1/LcrV antigens. Taking into account that capsule-negative Y. pestis strains are fully virulent, found in nature, or can be easily created, the LcrV is an essential component of the vaccine. Existing natural polymorphisms of LcrV [242,243] may require the addition of different variants of this antigen in vaccine formulations. Moreover, there are engineered versions of LcrV that avoid cross-protection [244]. Therefore, identification and inclusion of additional Y. pestis antigens, particularly antigen eliciting robust T cell response, should be an immediate priority for development of PSVs. There are several candidates with limited ability to elicit protective immunity, such as, YopD and YopE. The most reliable antigen is likely to be YscF, a polymeric subunit of the T3SS apparatus. The inclusion of YscF into adenovirus 5-based vaccine in addition to F1 and LcrV (Ad5-YscF-F1-LcrV) contribute to enhanced protection provided by this tri-valent PSV alone or in combination with purified antigens or LPV. This vaccine candidate provides a high level of protection against bubonic and pneumonic plague, even after a single dose, while both homologous and heterologous boosting confers 100% efficacy in different animal models [245–247]. Moreover, a hybrid combination YscF-F1-LcrV (YFV) in Ad5, in which the antigens are separated by a linker (GGGS) with the expression optimized for humans, can already serve as a prototype candidate for the mRNA vaccine.
Another important aspect to consider during vaccine design and testing is immune correlates of protection, particularly in humans, although thus far this issue has not been determined for candidate plague vaccines. This finding will likely be particularly important during optimization of PSV formulations, development of novel LPVs, or development of other types of vaccines, such as mRNA vaccines. The rubric defining the parameters of humoral and cellular immunity results of direct protection studies in different animal models, the longevity of immunity, and the evaluation of safety for the candidate plague vaccines remains to be determined for human use. Nevertheless, one quantitative characteristic was introduced for the testing of LcrV-based PSV. The LcrV protein is located at the tip of the T3SS needle guiding the injection of virulence effectors and impairing this function of LcrV disables the T3SS that is essential for virulence. The ability of anti-LcrV antibodies to block T3SS function can be evaluated with different reporters injected by T3SS to the host cell [248], although this approach has not been standardized. Moreover, LcrV is a multi-functional virulence factor that in addition to involvement with the translocation of effectors via the T3SS, contributes to immunomodulatory activity of Y. pestis [249,250]. Therefore, focusing strictly on titers of antibodies capable of diminishing T3SS translocation function may miss other important features of anti-LcrV antibodies related to the neutralization of immunomodulatory properties of secreted LcrV. The contribution of this part in protection against plague is unknown.
Overall, as outlined in recent WHO recommendations, plague vaccines should have no more than two doses of administration, provide long-lasting protection in both humoral and cellular immunity categories, not require cold chain storage, be injected by a needle-free method, and have robust safety profiles, including immunocompromised individuals [251]. While these criteria remain an aspiration, a prime-boost regimen involving LPV as the initial vaccination followed by PSV boost could represent an effective, rapid solution in case a significant plague outbreak suddenly occurs.
KNOWN 8: BIOSAFETY AND BIOSECURITY OF PLAGUE
Plague as a bioweapon
The earliest documented use of bioweapons date back to the middle of the 14th century. Between 1343 and 1347, the Genoese city, Caffa (now Feodosiya, Ukraine), was besieged by Mongol (Tartars) troops. An epidemic of plague struck the Tartars and caused severe mortality in their camps. The Tartars then catapulted their deceased plague-infected compatriots into Caffa, thus causing plague outbreaks in the city [252].
In the 1930s, a secret branch of the Imperial Japanese Army, known as the Kwantung Army Epidemic Prevention and Water Supply Department (known as Unit 731), developed Y. pestis as bioweapons. Clay vessels filled with infected fleas or Y. pestis-contaminated food was airdropped into populated areas in China with an attack on Ningbo City in 1940, killing 112 civilians [253].
On 10 April 1972, 78 nations signed the Convention on the Prohibition of the Development, Production, and Stockpiling of Bacteriological (Biological) and Toxin Weapons and on their Destruction (BWC). The enforcement of BWC legally terminated the offensive plans of bioweapons worldwide. Some countries, such as the former USSR, had earlier weaponized Y. pestis but the offensive bioweapon programs was officially closed in 1992 [254,255].
Y. pestis as a bioterrorism agent
After the anthrax letter attacks in the USA [256], pathogen-based bioterrorism has become an immediate threat to global security. Bioterrorism is defined as “the deliberate release of viruses, bacteria or other agents used to cause illness or death in people, animals, or plants. It is aimed at creating casualties, terror, societal disruption, or economic loss, inspired by ideological, religious or political beliefs” [257]. Contrary to biological warfare, in a bioterrorism attack biological agents are intentionally released against a civilian population.
The US Department of Health and Human Services and the Department of Agriculture have identified certain ‘Select Agents’ or ‘Biological Agents,’ which are pathogens and toxins with the potential to pose a severe threat to public health and safety. Because Y. pestis is easy to disseminate and transmit, and causes a high mortality rate in humans, Y. pestis is designated as a category A ‘select agent’ [258]. Y. pestis has been tagged as a high-priority agent, might be on the top of the list of potential bioterrorism agents, and could pose great risk to national and international biosecurity.
Biosafety regulations for Y. pestis
Several laboratory-acquired infection cases have been reported in the United States, some of which were fatal. The most recent lethal case was due to an attenuated strain, KIM D27, infecting a laboratory researcher. The condition of hereditary hemochromatosis coupled with diabetes in the researcher is believed to have contributed to the fatal course of the disease [259]. Occupational infections of plague have also been reported in veterinary staff, pet owners, and marmot hunters [260,261].
Due to the risk of laboratory-acquired infections approved local laboratory containment practices are recommended for all manipulations of suspect cultures, animal necropsies, and experimental animal studies. Characterized strains of reduced virulence, such as Y. pestis strain A1122, and KIM can be manipulated at lower containment levels [262]. These general recommendations in the U.S. Department of Health and Human Services are not fully accepted by other countries. For example, suspected plague samples in France are first dealt with at a lower containment level, but all culture work must be performed at a higher level. Avirulent and vaccinal strains with a long history could be handled in BSL1 with strict respect to the BSL1 regulation to avoid dissemination of the bacteria (i.e., lab coat, gloves, and overshoes).
UNKNOWN 8: RAPID RECOGNITION AND CONTROL OF PLAGUE IN DELIBERATE RELEASE OF Y. PESTIS
Deliberate release of Y. pestis
Theoretically, deliberate release of Y. pestis can be categorized as biological warfare, bioterrorism, and a biocrime. According to the 1972 BWC, nations are prohibited to undertake research to produce biological weapons or to produce and stockpile biological weapons. Although BWC has no inspection mechanism, it is unlikely that any sovereign nation would now pursue a bioweapon program. Y. pestis-related biocrimes have been documented, and in 1933 Dr. Taranath Bhatacharyna, a Calcutta physician with expertise in bacteriology, together with Benoyendra Chandra Pandey, injected a lethal dose of Y. pestis in the arm of Amarendra Pandey (the half-brother of Benoyendra Chandra Pandey). Three years thereafter both perpetrators were convicted and sentenced to death [258].
Possible ways of deliberate release of Y. pestis
The release of fleas infected with Y. pestis could be a viable method of releasing plague. Assuming this maneuver was successful, an attack with fleas would primarily cause bubonic plague [253]. Due to the low mortality rate of bubonic plague, infected fleas will not be an efficient way to deliberately release Y. pestis. Such an attack, however, might potentially generate a plague wild-life reservoir in a plague-free region and may result in long-term ecologic and economic consequences, in addition to being a public health issue.
Pneumonic plague-related dispersal would be an ideal way to intentionally spread Y. pestis. An aerosol release of 50 kg of Y. pestis over a city of 5 million people would result in 150,000 initial clinical cases and 36,000 deaths [263]. This previously-modelled scenario should be revisited according to our incremental experiences against epidemics and diagnostic and treatment tools in hands [264,265].
Another potential dispersal method for the deliberate release of Y. pestis would be through a suicide attacker that would lead to an explosion in cases of pneumonic plague originating from a single source or multiple sources with secondary infections. Without adequate precautions modern transportation systems could then readily deliver plague rapidly across the globe [266].
Countermeasures for deliberate release of Y. pestis
The most important issue combating deliberate release of Y. pestis is timely and effective responses of public health systems. Scientists are obligated to develop assays to identify wild-type and genetically-modified strains rapidly and accurately. The third-generation genome sequencing platform, such as NanoPore, will be of great value in this respect.
In the hypothetical scenario of the deliberate release of Y. pestis, timely and accurate recognition of the pathogen would be extremely difficult due to the overall complexity of the outbreak. In this case, a symptom surveillance system will be of help to identify potential patients as early as possible. Any unusual and clustered symptoms will trigger alerts and subsequent investigations and reduce the response time for combating the possible intentional release of Y. pestis.

