
1. BACKGROUND
In the three decades since the first gene therapy was performed, in 1990, the development of gene therapy has involved a challenging journey with many intricacies and complications. The gene therapy industry is currently valued at approximately 10 billion dollars per annum. Gene therapy, with its potential to radically change the treatment of viral infections, genetic diseases, cancer and many other conditions, has become one of the most studied and scrutinized pharmaceutical fields in recent years. More than 75,000 mutations have been linked to human ailments, almost half which are attributed to single-base mutations that cannot be cured by traditional therapies. In these intractable cases, lifelong medication may become necessary, thus increasing the risk of adverse effects and placing patients under psychological pressure. Consequently, a method for accurate and efficient correction of pathogenic point mutations is critical to enabling potential therapeutic approaches for genetic diseases. The discovery of CRISPR/Cas9 in 2012 paved the way to the development of such therapies. The subsequent refinement of this system has culminated in the development of base editing, an exceptional technology that is the most recent genome-editing technology available. Base editing has evolved rapidly and entered the clinical development phase, thus providing an auspicious new route for treating genetic disorders.
Base editing is a groundbreaking gene editing technique based on the CRISPR/Cas system developed by Liu and colleagues at Harvard in 2016 [1]. In contrast to CRISPR/Cas editing, base editing effectively modifies single-point mutations without generating DSBs. Because DSBs disrupt the nuclear structure and lead to the formation of chromosome bridges and micronuclei, which may eventually fragment chromosomes [2], base editing is more effective and safer than methods involving DSBs. Base editors (BEs) consist of three main components: a guide RNA (gRNA) that binds approximately 20 DNA nucleotides adjacent to the target base; a modified Cas9 enzyme that connects to the gRNA and nicks the DNA backbone on the strand opposite from the target base; and a base-editing enzyme, such as a deaminase, that makes precise chemical alterations to the desired DNA base or various bases simultaneously. After base substitution and DNA backbone nicking, the DNA repair mechanisms within the cell are activated and repair the nick while accurately pairing the new base with its complement, thus entirely replacing the original base on both strands [1].
Base editing combines the precision of the CRISPR machinery with the advantages of deaminase enzymes, thereby enabling genome modification without induction of DSBs ( Figure 1 ). Various types of BEs can produce specific base changes in DNA. Cytosine base editor (CBE) [1, 3], Adenine base editor (ABE) [4, 5] and guanine base editor (GBE) [6–8] convert cytosine to thymine (C→T), adenine to guanine (A→G) and cytosine to guanine (C→G), respectively. However, effective tools are needed for the other six possible base transitions (C→A, G→T, A→C, A→T, T→A and T→G), as well as base insertion and deletion. In 2019, Liu’s group introduced a versatile editor known as PE, which can perform all 12 base conversions and insertions/deletions without relying on DSBs or donor DNA [9–11]. Furthermore, mitochondrial BEs have also been developed to enable editing of mitochondrial DNA (mtDNA), which is not possible with other CRISPR-based BEs, such as ABE, CBE, GBE and PE [12–15].
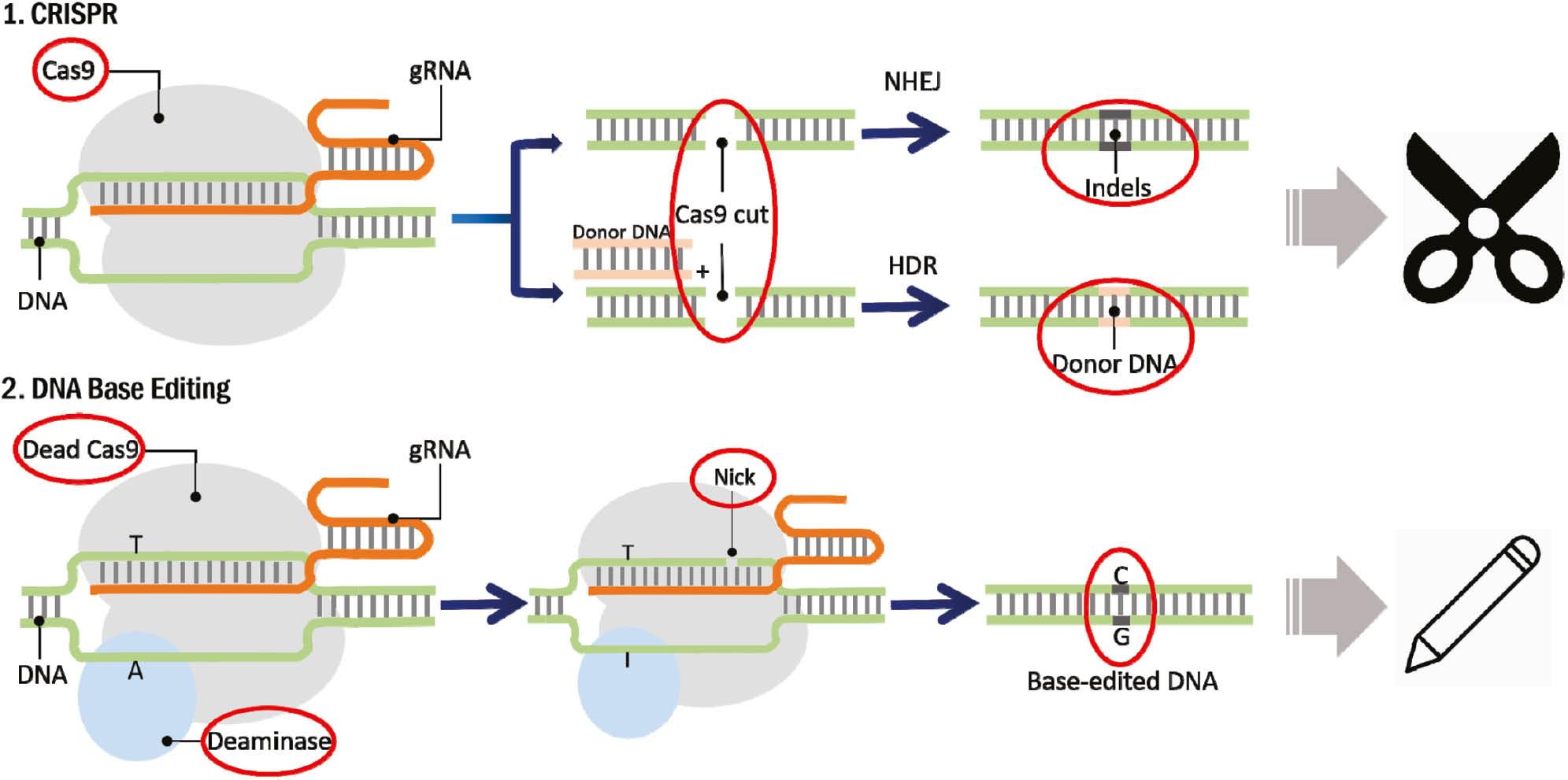
The schematic diagram of CRISPR and base editing.
CRISPR is like scissors for the genome, and BE is like a pencil for erasable writing. The red circle in the figure indicates the differences between these two techniques.
BEs enable accurate and efficient editing of a single-base substitution at a specific point in the genome without an associated risks of DSBs, thus decreasing the possibility of unintended chromosomal abnormalities and genotoxicity. Consequently, BEs are uniquely positioned to have excellent application value in ubiquitous point mutation editing.
BEs have a diverse range of potential applications in gene editing. First, they can be used to directly edit point mutations to restore gene functionality through a process referred to as gene correction. Second, BEs can be used to integrate protective clinical variants to mitigate or eliminate disease risk, in a process referred to as gene modification. Third, BEs can be applied to edit regulatory elements and reactivate gene expression, in a process known as gene activation. Fourth, BEs can be used to change specific nucleotide bases in a gene, thereby creating a stop codon to silence gene expression, or to edit splice sites to prevent the production of specific proteins, in a process referred to as gene silencing. Finally, BEs can be used for multiplex editing, in which multiple sites are edited simultaneously with no detectable translocations.
2. CLINICAL TRIALS AND PRECLINICAL STUDIES
Base editing therapy has advanced to the clinical development stage, and various clinical trials are currently in progress to treat diseases such as sickle cell disease (SCD), β-thalassemia, familial hypercholesterolemia and universal chimeric antigen receptor T-cell (CAR-T) therapy. Details of clinical trials ( Table 1 ) and preclinical studies ( Table 2 ) are described below.
Clinical trials of base editing therapies.
| Study title | ClinicalTrials.gov identifier | Intervention | Targets | Way of action | Conditions | Phase |
|---|---|---|---|---|---|---|
| BEACON: A Study Evaluating the Safety and Efficacy of BEAM-101 in Patients With Severe Sickle Cell Disease | NCT05456880 | BEAM-101 [16] | HPFH single nucleotide polymorphism | CD34+ HSPCs | SCD | Phase 1/2 |
| A Study of VERVE-101 in Patients With Familial Hypercholesterolemia and Cardiovascular Disease | NCT05398029 | VERVE-101 [17] | PCSK9 | LNP, in vivo | HeFH, ASCVD and hypercholesterolemia | Phase 1 |
| Study of Base Edited CAR7 T Cells to Treat T Cell Malignancies (TvT CAR7) | NCT05397184 | BE CAR7 [18] | TCR, CD7 and CD52 | CAR-T cells | R/R T-cell ALL | Phase 1 |
| NA | IND | BEAM-102 [19] | S point mutation | CD34+ HSPCs | SCD | Na |
| A Study Evaluating the Safety and Efficacy of BEAM-201 in Relapsed/Refractory T-cell Acute Lymphoblastic Leukemia (T-ALL) or T-cell Lymphoblastic Lymphoma (T-LL) | NCT05885464 | BEAM-201 [20] | CD7, TRAC, CD52 and PD-1 | CAR-T cells | R/R T-ALL and CD7+ AML | Na |
| NA | IND | BEAM-301 [21] | R83C mutation in the G6PC gene | LNP, in vivo | GSDIa | Na |
| NA | IND | BEAM-302 [22] | E342K mutation in the SERPINA1 gene | LNP, in vivo | AATD | Na |
SCD, sickle cell disease; HeFH, heterozygous familial hypercholesteremia; ASCVD, atherosclerotic cardiovascular disease; R/R T-cell ALL, relapsed/refractory T-cell acute lymphoid leukemia; TDT, transfusion-dependent β-thalassemia; GSDIa, glycogen storage disease 1a; AATD, alpha -1 antitrypsin deficiency.
Preclinical studies on base editing therapies.
| BE | Targets | Way of action | Conditions |
|---|---|---|---|
| ABE/CBE | HBG promoter [23] | CD34+ HSPCs | TDT and SCD |
| ABE8e V106W | Codon 26 of the HBB gene [24] | Mice | HbE β-thalassaemia |
| ABE8e | BCL11A enhancer or HBG promoter [25] | CD34+ HSPCs | TDT and SCD |
| ABE8e-SpRY | HbE and IVS II-654 mutations [25] | CD34+ HSPCs | β-thalassaemia |
| CBE | CD52, CD7, PD1 and TCRα [20] | 7CAR8 multiple in vitro and in vivo models | T-cell leukemia and other CD7+ malignancies |
| CBE (SpCas9-NG) | Hemophilia B (c.947T>C; I316T) [26] | IPSCs TK-NOG mice | Hemophilia |
| ABE | CD3D [27] | HSPC mice | CD3δ SCID |
| ABE | c.1208G>A (p.R403Q) in β-myosin [28] | IPSC cardiomyocytes | HCM |
| PE | ABCA4 [29] | Cell lines | Stargardt’s disease |
| ABE | Z mutation in SERPINA1 [30] | IPSCs iHeps | AATD |
| PE | E342K mutation (G to A) in SERPINA1 (PiZ allele) [31] | AAV, mouse liver | AATD |
| CBE | Integrated HBV DNA and covalent closed circular DNA [32] | Mice | HBV |
| ABE | LMNA [33] | Patient fibroblasts; AVV, mouse model | HGPS |
| dCas13X.1-ADAR2ddE488Q (mxABE) | Myosin VI p.C442Y [34] | AAV-PHP, mouse model | Autosomal dominant hearing loss |
TCRα, T-cell receptor α chain; PD1, programmed cell death protein 1; SCID, severe combined immunodeficiency; HCM, hypertrophic cardiomyopathy; AATD, a-1 antitrypsin deficiency; HGPS, Hutchinson-Gilford progeria syndrome.
2.1 DNA base editing
2.1.1 In vitro therapies
β-thalassemia and SCD are the most prevalent monogenic diseases worldwide. Both diseases can be cured through allogeneic or genetically engineered hematopoietic stem cell (HSC) transplantation. In patients with β-thalassemia and SCD with hereditary persistence of fetal hemoglobin (HPFH), naturally occurring point mutations in the HBG promoter can switch hemoglobin synthesis from defective adult β-globin to fetal γ-globin, thus potentially ameliorating the clinical manifestations of these hemoglobinopathies. Researchers inspired by the natural phenomenon of HPFH have endeavored to induce similar mutations through base editing. Specifically, researchers have leveraged base editing to induce mutations in the HBG promoter, thus resulting in the upregulation of fetal hemoglobin (HbF) production in erythroblasts derived from human CD34+ hematopoietic stem and progenitor cells (HSPCs), to increase the therapeutic potential of HbF [25, 35, 36]. These mutations either create new binding sites for erythroid activators, such as TAL1, KLF1 and GATA1 (−175T>C, −198T>C and −113A>G) [37–39], or disrupt the binding sites of repressors, such as BCL11A and ZBTB7A/LRF (−114C>A, −117G>A and a 13 bp deletion [Δ13 bp], −195C>G, −196C>T, −197C>T, −201C>T and −202C>T/G) [40]. As a result of these mutations, the expression of HbF significantly increases, thus offering promising therapeutic benefits for patients with hemoglobinopathies. Furthermore, other mutations have been reported. Mohankumar et al. have demonstrated in vitro that a de novo binding site for KLF1 can be created via the introduction of −123T>C and −124T>C HPFH-like mutations, thereby driving γ-globin expression [23]. Davies et al. have described a base-editing strategy for hemoglobin E (HbE) β-thalassaemia, in which the HbE mutation is restored to either its wildtype form or to a normal variant hemoglobin (E26G), also known as Hb Aubenas. This approach has resulted in the recreation of an asymptomatic trait phenotype in NGS mice [24]. Similarly, Wu et al. have successfully applied the ABE8e variant, an ABE, to introduce nucleotide substitutions in the BCL11A enhancer or HBG promoter. These substitutions upregulate γ-globin expression in CD34+ HSPCs from patients with β-thalassemia. Moreover, a near-PAMless ABE variant, called ABE8e-SpRY, has been developed and used to correct HbE and IVS II-654 mutations in patient-derived CD34+ HSPCs. This intervention has resulted in durable therapeutic editing in self-renewing repopulating human HSCs, as assayed in primary and secondary recipients [25]. Together, these results support the potential of base editing in HSCs as a therapeutic approach for treating inherited monogenic blood disorders.
Early clinical trials on base editing techniques for the treatment of β-thalassemia and SCD were conducted by Beam Therapeutics, a leading biotechnology company specializing in base editing. BEAM-101 is an in vitro HSC therapy for the treatment of SCD and transfusion-dependent β-thalassemia, whereas BEAM-102 is intended for the treatment of SCD alone. Both therapies use electroporation to introduce base editing tools into cells obtained from a patient’s body. BEAM-101 aims to replicate the single nucleotide polymorphisms present in HPFH to combat the effects of mutations causing SCD or β-thalassemia. Preclinical studies have indicated that BEAM-101 produces a high degree of HSC editing (in more 90% of alleles), thus significantly increasing HbF to levels accounting for more than 60% of the total hemoglobin and markedly decreasing the expression of the pathogenic protein HbS, to levels similar to those in SCD carriers without the disease [19]. The FDA has approved the IND for BEAM-101, and the BEACON trial (NCT05456880), examining the safety and efficacy of this therapy in treating patients with severe SCD, is currently underway. In contrast, BEAM-102 is an investigational therapy designed to modify the hemoglobin S causative point mutation and create a naturally occurring, normal human hemoglobin variant known as HbG-Makassar, which is believed to be a natural functioning hemoglobin variant.
In May 2022, a 13-year-old girl named Alyssa, with T-cell acute lymphoblastic leukemia (T-ALL), became the first patient to receive base editing allogeneic CAR-T therapy. This breakthrough came 10 years after a girl named Emily became the first cancer patient to receive CAR-T therapy and achieve clinical cure. The allogeneic CAR-T cells underwent four steps of genetic modification. First, the TCR gene was knocked down to decrease the risk of graft-versus-host disease. Second, the T-cell marker gene CD7 was eliminated to prevent fratricide of the donor T-cells. Third, CD52 was knocked down with base editing to enhance the modified T-cells’ resistance to chemotherapy. Finally, a CAR targeting CD7 on leukemic T-cells was added to the genetically modified T-cells [41]. Qasim et al. are conducting a clinical trial of BE Car7-T cell therapy TvT CAR7 (ISRCTN15323014), in a non-randomized single-arm open-label single-center clinical trial aimed at enrolling ten participants [42]. The goal of the TvT CAR7 study was to assess the safety and feasibility of BE-CAR7 in treating children with T-ALL before HSC transplantation. Early clinical efficacy results from the TvT CAR7 study were presented at ASH2022 [43]. After 28 days of treatment, Alyssa achieved remission and underwent a bone marrow transplant to restore her immune system. Six months after treatment, her condition had significantly improved, and her cancer cells were undetectable [18, 43].
On December 2, 2022, the FDA approved Beam Therapeutics’ base editing-based CAR-T cell therapy, BEAM-201, for use in a clinical trial addressing relapsed/refractory acute T-lymphoblastic leukemia (R/R T-ALL) and T-lymphoblastic lymphoma. BEAM-201 is a CD7-targeting CAR T therapy that uses base editing to disable four genes: CD7, TRAC, CD52 and PD-1. The therapy is tailored to improve the long-term anti-tumor activity of CAR T cells with respect to that of the treatment received by Alyssa. Beam Therapeutics is also developing another multi-gene edited CAR-T therapy for T cell lymphoma, which targets CD5 and remains unnamed.
Moreover, a preclinical study led by Diorio et al. in August 2022 used base editing to formulate a potent and complex gene-edited CD7-specific CAR-T product (7CAR8) for off-the-shelf use in patients with T-cell leukemia and alternative CD7+ malignancies. The outcomes provide cause for optimism and a strong justification for further clinical application [20].
Researchers have recently demonstrated the successful conversion of a mutation from C to T in induced pluripotent stem cells (iPSCs) from a patient with hemophilia B (c.947T>C; I316T), by using a base editing approach incorporating SpCas9-NG. The edited iPSCs were differentiated into hepatocyte-like cells in vitro and, after subrenal capsule transplantation into TK-NOG mice, expressed notable levels of F9 mRNA. Furthermore, SpCas9-NG-mediated base editing restored the production of coagulation faction in both HEK293 cells and knock-in mice by correcting the mutation [26], thus suggesting potential therapeutic value in treating genetic diseases such as hemophilia B. These findings underscore the efficacy of the base editing approach using SpCas9-NG’s broad PAM flexibility, as well as its promise as a viable solution in combating genetic diseases.
CD3δ severe combined immunodeficiency (SCID) is a debilitating immunodeficiency caused by mutations in CD3D, a gene encoding the invariant CD3δ chain, which is essential for normal thymopoiesis, in the CD3/TCR complex [44]. Kohn et al. have recently used an ABE strategy to restore CD3δ in patient-derived autologous HSPCs. Delivery of mRNA encoding ABE and gRNA resulted in a 71.2% ± 7.85% (n=3) rate of correction of the pathogenic mutation. The edited HSPCs differentiated into artificial thymic organoids that produced mature T cells with diverse TCR repertoires and TCR-dependent functions. Edited human HSPCs transplanted into immunodeficient mice showed an 88% rate of reversion of the CD3D defect in human CD34+ cells extracted from mouse bone marrow after 16 weeks, thus indicating that long-term HSC (LT-HSC) had been corrected. These findings demonstrate the effectiveness of ABE in HSPCs in treating CD3δ SCID, thereby paving the way to the potential development of a one-time treatment for patients with CD3δ SCID [27].
Hypertrophic cardiomyopathy (HCM) is a frequently encountered disease of the heart caused by cardiac sarcomeric gene variants and resulting in anomalous thickening of the heart muscle. One particularly prevalent pathogenic variant is c.1208G>A (p. R403Q) in β-myosin, which promotes HCM onset by intensifying cardiac contractility. Recently, several studies have reported successful remediation and amelioration of HCM phenotypes in patient-derived iPSC cardiomyocytes by using an ABE-mediated strategy. Encouragingly, the feasibility of base editing for treating hereditary cardiac diseases has been demonstrated by promising results in a humanized mouse model of HCM [45].
Stargardt’s disease is caused by a single-base mutation in the ABCA4 gene. This autosomal recessive genetic disorder manifests as yellow spots in the fundus and steadily decreasing central vision. ABCA4 has approximately 1,200 known pathogenic mutations, approximately 63% of which are transition mutations. Recent data obtained from gnomAD, Leiden Open Variation Database (LOVD) and ClinVar, screened for PAM sites in relevant BEs, have revealed that 53% (ClinVar), 71% (LOVD) and 71% (gnomAD) of these potentially editable mutations are pathogenic transition mutations. Among these, 35–47% have PAM sites considered “ideal.” Moreover, 16–20% of these mutations are located within a range of multiple PAM sites, thus enabling various editing strategies to be tested. Recent research has indicated that prime editing is highly effective in correcting mutations in the porcine ABCA4 gene, thus emphasizing the importance of appropriate ngRNA design and offering a promising avenue for treating Stargardt’s disease through gene therapy [29]. These studies have provided a clearer understanding of the potential for base editing approaches as gene therapy strategies for Stargardt’s disease [46, 47].
Alpha -1 antitrypsin deficiency (AATD) is a common inherited disorder that is caused by a single-base pair Z mutation in the SERPINA1 gene, and leads to chronic and progressive lung and liver disease. Recently, ABE has been used to correct the mutation in induced iPSCs and iPSC-derived hepatocytes (iHeps) from patients with AATD. Decreased aberrant AAT accumulation and increased secretion in patient iPSCs, as well as minimized ER stress in iHeps, were observed. The efficiency of ABE in iPSCs has been determined to be highly efficient (95%) with respect to that of CRISPR/Cas9 (0.3%). Moreover, whole-genome sequencing of treated cells has indicated no off-target genomic mutations [30]. In 2021, Liu et al. demonstrated the practicability and effectiveness of base editing by using dual AAVs for delivering split-intein NLS-optimized SpCas9-based PE to rectify the E342K mutation (G to A) in SERPINA1 (PiZ allele) in the mouse liver [31]. These findings underscore the viability of base editing as a reliable tool for correcting the Z mutation in cells from patients with AATD.
2.1.2 In vivo therapies
Although in vitro editing is feasible for some critical cell types, such as HSCs, several other cell types are not amendable to this technique. Hence, in vivo gene editing therapies are emerging as a potential solution to treat the root causes of various genetic disorders [48].
Safe and efficient delivery of editing systems to target cells is critical and is a major challenge in the therapeutic success of BEs. The most common delivery system used for base editing is lipid nanoparticle (LNP) technology. This method has shown tremendous promise in the delivery of the novel coronavirus mRNA vaccines approved by the FDA in 2020. This achievement has substantially advanced the development of LNP technology as a promising delivery system. Another frequently used vector is the adeno-associated virus (AAV), a small virus belonging to the parvovirus family, with a size slightly above 20 nm, approximately equivalent to two or three antibody molecules. Currently, hundreds of AAV gene therapy products are undergoing clinical trials for various indications, and AAV gene therapy has made major strides and reached an impressive stage of progress.
On May 19, 2021, a preclinical study demonstrated the potential of LNP-delivered ABE to accurately edit the PCSK9 gene splicing sites in non-human primate models. The treatment effectively decreased the levels of PCSK9 and low-density lipoprotein cholesterol (LDL-C) in the blood. Two weeks after injection, the expression of the PCSK9 gene in the liver cells of monkeys had decreased by 67%, the PCSK9 protein in the blood had decreased by 89%, and levels of LDL-C had decreased by 61%. Importantly, the gene editing therapy exhibited no off-target or toxic effects [49]. This proof-of-concept demonstration provided the first indication that single-base editing in the liver in non-human primates can achieve long-lasting lowering of LDL cholesterol to prevent cardiovascular disease.
In another preclinical study conducted on October 31, 2022, the safety, efficacy, persistence, tolerability, and effects on germ cells were evaluated after editing PCSK9 with Verve-101 therapy in monkeys. The findings showed that monkeys receiving a dose of 0.75 mg/kg had, on average, 67% less PCSK9 protein and 49% less LDL-C in their blood 1 year after than before treatment, whereas those receiving a dose of 1.5 mg/kg had an average 83% decrease in blood PCSK9 protein and a 69% decrease in LDL-C after 476 days of treatment. Additionally, editing of PCSK9 was not detected in the sperm of male monkeys after base editing treatment or in the offspring of female mice after base editing treatment [17]. These results further support the potential of base editing for the safe and effective treatment of PCSK9-associated cardiovascular disease.
In July 2022, a phase I clinical trial (NCT05398029) for Verve-101, a BE based therapy designed to treat heterozygous familial hypercholesteremia (HeFH) commenced. This clinical study is the first in vivo BE therapy to enter the clinical development stage. The first patient administration has been successfully completed in New Zealand. Enrollment of 40 patients is planned, and VERVE-101 will be delivered by LNP to the patients’ liver cells. The therapy works by permanently switching off the expression of the PCSK9 gene through a single-base A-to-G substitution at a specific site in the PCSK9 gene, thereby decreasing LDL-C levels. This innovative approach can prevent or treat LDL-C related cardiovascular diseases, including HeFH [17].
Chronic hepatitis B virus (HBV) infection remains a major global health burden, and current standard therapies predominantly involve nucleotide/nucleoside analogues (NUCs) and interferons. NUCs inhibit only viral DNA replication and require long-term medication, thus resulting in functional cure in a mere 3% of patients.
In September 2022, Beam Therapeutics presented a study on the use of CBE technology to target and interrupt HBV replication. CBE technology significantly decreased the levels of HBV surface antigen (HBsAg) and other viral biomarkers. To achieve these results, the researchers targeted multiple genomic sites in HBV-infected cells to precisely and permanently introduce stop codons into the integrated HBV DNA and covalently closed circular DNA without a risk of chromosome rearrangement. Consequently, HBV-associated markers such as HBsAg, HBeAg, HBV DNA and 3.5 kb RNA substantially decreased. In an HBV-infected mouse model, one to two doses of base editing therapy (mRNA and gRNA of CBEs delivered by LNPs) led to a sustained decrease in HBsAg, by >2 log10 IU/ml, and a sustained decrease of 3 log10 copies of HBV DNA per ml serum.
A report has described the successful use of SpCas9-derived BE to edit episomal covalently closed circular DNA and suppress viral gene expression in an in vitro HBV infection system [50]. In another study, CBE has been used to introduce a stop codon in the HBV S gene. A premature stop codon in the S gene was introduced in 71% of human hepatoma PLC/PRF/5 cells carrying the HBV genome. These findings demonstrate the potential of base editing therapy as a candidate approach for eradicating HBV [32].
These studies underscore the potential of base editing therapy in treating and eliminating HBV. Furthermore, the multi-base editing approach is a promising avenue for developing gene therapies for treating HBV and other chronic viral infections.
Glycogen storage disease 1a (GSDIa) is a metabolic disorder caused by congenital enzyme defects that affect glycogen metabolism. This condition is often associated with genetic mutations and follows an autosomal recessive inheritance pattern. BEAM-301 is a base editing therapy that targets the R83C mutation in the G6PC gene and is aimed at treating GSDIa through LNP-mediated delivery of mRNA of the base editing components to the liver. Preclinical studies have demonstrated high and sustained editing efficiency for as many as 35 weeks in a mouse model of GSDIa [21].
On January 6, 2021, a study led by the Liu and Brown groups was published online in Nature. The study used ABE to directly correct pathogenic mutations associated with Hutchinson-Gilford progeria syndrome (HGPS) in fibroblasts from children with progeria and a mouse model of HGPS. The results demonstrated no off-target editing. To assess the efficacy of this base editing approach, the team worked with the Progeria Research Foundation to deliver ABE via lentivirus to laboratory fibroblasts from children with premature aging to edit their LMNA gene. The mutation was successfully corrected in a site-directed manner in 87% to 91% of cells. Moreover, the researchers further tested this gene editing strategy in a homozygous transgenic mouse model of premature aging (C57BL/6-tg (LMNA*G608G) HClns/J) by using AAV9 to deliver ABE via a single injection. The results demonstrated successful restoration of the normal DNA sequence of the LMNA gene in many cells in different organs, including the heart and aorta. Most cell types exhibited the correct DNA sequence 6 months after treatment. In the aorta, the edited cells were observed to have replaced the cells carrying the premature senescence mutation, which degenerated due to early deterioration. Critically, a single injection of AAV9-ABE at postnatal day 14 improved viability, increasing the lifespan of treated mice from 7 months to almost 1.5 years. The normal lifespan of the mouse model used in the study was typically 2 years. These findings collectively demonstrate that in vivo base editing strategies have high potential as alternative treatment options for HGPS and other genetic disorders by accurately correcting the root cause [33].
2.1.3 MtDNA base editing
Mitochondria serve as cellular energy factories and contain a distinct genetic material known as mtDNA. Single base mutations can substantially affect mtDNA and cause a range of disorders, including neuromuscular disorders, developmental diseases, cancer and other illnesses. Unfortunately, current methods are unable to efficiently introduce foreign RNA into mitochondria, thus rendering CRISPR-based gene editing tools useless for mitochondrial gene editing.
However, Liu’s laboratory has discovered DddA, an interbacterial toxin mediating the deamination of double-stranded DNA cytidine, and used it to develop the first DddA-derived cytosine base editor (DdCBE) independent of CRISPR, in July 2020 [13]. In May 2022, Kim’s team developed transcription-activator-like effector (TALE)-linked deaminases (TALEDs), which induce targeted A-to-G editing in human mitochondria [12]. With the rapid advances in mtDNA editing tools, this innovative technique appears poised to be tested in clinical settings soon.
DdCBEs [12, 13, 15] and TALEDs [12] enable programmable C•G-to-T•A and A•T-to-G•C conversions in mtDNA, respectively, without a need for DSBs. They could potentially model or correct pathogenic variants implicated in mitochondrial disease, and enhance understanding of mitochondrial biology. Remarkably, analysis of the MITOMAP database [51] has revealed that 86 of 91 confirmed point mutations associated with disease in mtDNA could theoretically be corrected or modeled with DbCBEs or TALEDs.
2.2 RNA base editing
Beyond DNA editing, base editing can also target RNA. Adenosine deaminases acting on RNA (ADARs) can convert adenosine to inosine in double-stranded RNA [52]. In 2017, Zhang and colleagues developed a programmable adenosine-to-inosine (A-to-I) RNA editing strategy by combining catalytically inactive RNA-targeting CRISPR-Cas13 (dCas13) with the adenine deaminase domain of ADAR2 [53]. Subsequently, in 2019, they reported RESCUE, a cytidine-to-uridine (C-to-U) RNA editor, by directly evolving ADAR2 into a cytidine deaminase [54]. In August 2021, they discovered a novel RNA BE based on ultra-small Cas13bt nucleases, which can introduce C-to-U and A-to-I mutations. This newly developed RNA BE has an advantage of small size enabling delivery via a single AAV carrier system [55]. Additionally, in March 2022, AIMers, an ADAR, was reported as a small, chemically modified oligonucleotide editing tool that facilitates RNA base editing technology with potential for broader liver therapeutic applications in non-human primates [56]. AIMers corrects single-base mutations in RNA transcription, thereby avoiding the persistent changes to the genome that can arise from DNA-targeting methods. AIMers uses the ADAR enzyme to transform A to I, which is interpreted as G by cells, thus avoiding the need for complicated delivery vectors, such as viral vectors or LNPs, to achieve sustained editing in the liver, central nervous system and other tissues. AIMers can edit as much as 50% of the ACTB (β-actin) transcripts in non-human primate livers, with editing levels sustained at 40% or more for more than 1 month. These advances have considerably improved the applicability of RNA BEs in gene therapy.
2.2.1 Autosomal dominant hearing loss
In July 2022, Xiao et al. published research on a mini dCas13X.1-based adenosine base editor (mxABE), which involves truncated Cas13X.1 and the RNA editing enzyme adenosine deaminase acting on RNA 2 deaminase domain variant (ADAR2ddE488Q); their findings have demonstrated both high A>G conversion efficacy and a low off-target editing frequency [34]. In this study, the authors evaluated the therapeutic potential of mxABE-mediated RNA correction in a Myo6C442Y/+ mouse model exhibiting the characteristics of human dominant-inherited deafness. Delivery of mxABE to the cochlea of the mice via AAV-PHP prevented the loss of cochlear hair cells while restoring hearing ability in the mice for 3 months post-injection [34].
3. SUMMARY
Base editing technology was first proposed in 2016 and has remarkably advanced in the following 7 years and culminated in human treatments. Clinical trials of base editing technology have begun for a variety of conditions, including leukemia, SCD, β-thalassemia and familial hypercholesterolemia ( Table 1 ).
Compared with traditional CRISPR gene editing techniques, base editing enables precise modification of a single site without breaking double stranded DNA. Nevertheless, the development of innovative drugs is fraught with challenges. Because the technology still carries a risk of off-target effects, safety is a major concern during the clinical development phase. The FDA’s clinical hold and suspension reflect the cautious approach to base-editing therapies.
Overall, the development of in-vivo base editing therapies has encountered several obstacles. Future breakthroughs are expected to focus on decreasing off-target effects, broadening the targeting range, and enhancing in vivo editing and delivery efficiency. In terms of clinical applications, safeguarding safety is the primary consideration. The effectiveness and delivery efficiency of gene editing therapy are also crucial factors to be considered when developing innovative drugs.
Compared with DNA editing, RNA base editing induces solely transient modifications in mRNA transcribed from DNA, thus avoiding any heritable or enduring off-target mutations, and mitigating potential safety and ethical concerns. Recent studies on RNA base editing have shown a growing interest in this innovative technique in the realm of drug development. As editing efficacy and safety continue to improve, researchers are exploring the development of simpler RNA base editing strategies. Looking ahead, we anticipate that an increasing number of innovative drugs in the base editing field will advance to clinical trials as the applications continue to expand. With time, we believe that base editing therapy will mature, and regulators, healthcare professionals and patients will gain a better understanding of its principles, therapeutic efficacy and drug safety. This improved comprehension will lead to greater acceptance of this novel therapy. We expect base editing to play a major role in expanding treatment options for patients and curing more diseases.