
1. INTRODUCTION
Lymphoid neoplasms are broadly divided into B-cell lymphoid, T-cell, NK-cell, and dendritic cell neoplasms, and Hodgkin lymphomas (HL) [1–3]. In contrast to the cure rate of ≥ 85% in patients (pts) with HL [4–6], the morbidity and mortality of adult non-Hodgkin lymphoma (NHL), including 45.8% of diffuse large B cell lymphoma (DLBCL), ranked 10th death in China before 2016 [7, 8]. Among more than 18 different DLBCL subtypes, worse outcomes were reported with co-occurrence of MYD88 L265P and CD79B mutation in activated B-cell-like (ABC)-DLBCL (MCD subtype), and ABC-DLBCL with gain-of-function of NOTCH1 mutation (N1 subtype). MCD and N1 subtypes had 26% and 36% 5-year overall survival (OS) after rituximab-based chemoimmunotherapy, respectively [9]. In addition, high-grade-B cell lymphomas with c-Myc/8q24 and BCL2/18q21 translocation (double hit) or with BCL6/3q27 translocation (triple hit) are highly resistant to chemotherapy [10]. Before the advent of autologous CD19 chimeric antigen receptor (CAR)-T cell (19CAR-T cell) therapy, the 2-year OS of patients with chemotherapy-resistant DLBCL was 20%, and the median OS was 6.2 months, according to the SCHOLAR-1 study [11]. Axicabtagene ciloleucel (axi-cel), prototypic 1928ζ CAR-T cells, have been reported to result in a 54% 2-year OS in patients with r/r DLBCL after two or more lines of treatment in the ZUMA-1 study [12, 13]. In addition, the introduction of axi-cel into second-line therapy in patients with r/r DLBCL or infusion of axi-cel as part of first line therapy in patients with double- or triple-hit HGBCL significantly increased event-free-survival (EFS), and overall response rate (ORR)/complete response (CR) compared with standard therapy [14, 15].
Despite high treatment effectiveness, grade ≥3 cytokine release syndrome (CRS) and immune cell associated neurotoxicity (ICAN) from axi-cel occur in 13% and 28% of treated patients with r/r DLBCL, respectively, after two or more lines of treatment [16–18]. Notably, CRS increases rapidly within 5–10 days after transfusion of axi-cel [17]. The clinical and biological factors of individual patients have been associated with the risk of CRS and ICAN after 19CAR-T cell therapies. These factors include individual inflammatory status, intestinal microbiota, tumor burden, genomic instability of r/r DLBCL, lymphocyte depletion protocols, intrinsic quality characteristics of 19CAR-T cells, pyroptosis of B-cell cancer, endothelial or macrophage activation, and IL-6 level [19–23]. During CRS progression, local (L)-CRS is observed in patients with a high tumor burden, including local activation and expansion of 19CAR-T cells, activation of myeloid cells [24], and secretion of tumor necrosis factor (TNF)-α, interferon γ, and IL-1ß. L-CRS is a precursor of systemic (S)-CRS [25]. S-CRS is ameliorated by synergistic blockade of TNF-α, IL-1ß, and inhibition of focal adhesion kinase (FAK) [26]. In addition, tocilizumab, corticosteroids, and ruxolitinib are frequently used to control S-CRS [27–29]. In addition, metoprolol, a cardioselective ß1-adrenergic receptor blocker, has recently been reported to improve CRS and serum levels of IL-6 [30].
Approximately 57% of patients with r/r DLBCL experience disease progression or relapse after autologous 19CAR-T cell therapy [31, 32]. For example, early disease progression within 30 days or after 30 days has been reported to have a median OS of 3.75 months and 9.28 months, respectively [33]. Tumor intrinsic factors, such as two or more extra nodal sites, a high total metabolic volume, elevated serum C-reactive protein (CRP) and lactate dehydrogenase, the presence of apolipoprotein B mRNA editing catalytic polypeptide 3 (APOBEC), cytidine deaminase mutagenesis-associated signatures 2 and 13, the presence of 3p21.31, a chromosomal deletion containing the tumor suppressor RHOA, aberrant apoptotic signaling, and oxidative DNA damage are closely associated with early disease progression after 19CAR-T cell treatment [34, 35]. In addition, CD19 loss and/or decrease in B-cell cancer is associated with resistance 19CAR-T cell therapy [36]. The mechanisms of CD19 loss in B-cell malignancies have been extensively studied [36]. These mechanisms include pre-existing CD19-negative clones [37], acquired genetic point mutation of CD19 exon 3 from p.163 (R to L) or p.174 (L to V) [38], loss of serine/arginine-rich splicing factor 3 (SRSF3) [39], hyper-glycosylation of CD19 [40], myeloid lineage switch due to dysregulation of PAX5 [41], Ikaros DNA binding protein [42], and the EBF1 zinc finger containing transcription factor [43]. Moreover, the presence of fewer than 3000 membrane bound CD19 molecules per r/r DLBCL results in failure to activate 1928ζ CAR-T cells. This aspect is reflected by decreased phosphorylation of extracellular signal-regulated kinase (pERK) in 19CAR-T cells [44, 45]. Furthermore, 19CAR-T cell exhaustion under an immunosuppressive tumor microenvironment (TME) leads to disease progression or relapse. Chronic TAA exposure in the TME results in constitutive activation of 19CAR-T cells, differentiation, epigenetic alteration, and upregulation of immune checkpoint molecules [46–48]. Recently, numerous novel strategies have been developed to modify T cells to overcome TME and tumor heterogeneity TAA. This literature review provides an overview of modular models of prototypical CAR constructs, and their synergistic effects in controlling T-cell function. Subsequently, novel CAR modifications and T-cell engineering strategies are summarized. Notably, bispecific tandem or loop CAR-T cells significantly enhance therapy-associated safety and high efficacy in patients with r/r DLBCL. In addition, CAR integrating at TCRα chain constant region by using clustered regularly interspaced short palindromic repeats (CRISPR) derived single guide RNA and Cas9 and recombinant adeno associated viral vectors (rAAV) enhances tumor rejection, and delays CAR-T cells exhaustion in vivo. Furthermore, human leukocyte antigen independent synthetic TCR and antigen receptors are able to respond to low density tumor antigens with little tonic signaling. Armored CAR-T cells facilitate CAR-T cell migration and infiltration, and survive in the TME of r/r DLBCL.
2. CAR ELEMENTS AND THEIR SYNERGISTIC EFFECTS IN CONTROLLING CAR-T CELL FUNCTION
2.1 Affinity and avidity of CAR
The B cell response to antigens depends on the antigen/BCR affinity threshold [49]. Activation of TAA specific CAR-T cells depends on the affinity of CAR, surface CAR density, and TAA expression per cancer cell [50–52]. Typical second-generation CAR constructs have had great success in treating hematologic cancers [53–55]. Second-generation CARs consist of five modular domains: the extracellular TAA binding domain, ectodomain of the hinge region, transmembrane domain, co-stimulatory domain, and three repetitive immune tyrosine activation motifs (ITAM) of the CD3ζ chain. The ectodomain of the antigen binding domain, together with the hinge region, facilitates TAA recognition. The second-generation CAR construct was developed from the first-generation antibody-based CAR construct, designed in 1989. The single chain variable fragment (scFv) of mouse monoclonal antibodies (mAb) against TAA is commonly used as a TAA binding domain. The development of camel heavy-chain antibody, human Fab antibodies, and natural immune receptors, such as NKG2D, has expanded the number of disease targets [56]. At the molecular level, scFv consists of a variable heavy chain (VH) and a variable light chain (VL). Each VH and VL has four framework regions (FR1–4) and three complementarity determining regions (CDR1–3). Specifically, CDR1–3 of CAR-scFv (FMC63 mAb, IgG2a) bind membrane-bound epitopes adjacent to exons 3 and 4 of CD19 [28]. FR1–4 of CAR-scFv facilitate binding of CDR1–3 to CD19. Studies have shown that FR1–4 of scFv of FMC63 mAb are much more stable than FR1–4 of scFv of GD2 specific CAR-T cells and CD22 specific BBζ CAR-T cells. Therefore, 19BBζ CAR-T cells have little antigen-independent scFv-clustering that triggers tonic signaling. In general, CAR-T cell tonic signaling results in TAA independent constitutive CD3ζ activation, terminal differentiation, and exhaustion of CAR-T cells [57, 58]. However, tonic signaling delivers activation signals to CD22BBζ CAR-T cells (m971 mAb) and overcomes insufficient reactivity against low-density CD22 TAA in r/r B-ALL. For CD22BBζ CAR-T cells, short scFv linkers (5 amino acids) have a much more autonomous tonic signaling effect through antigen-independent multimerization of scFv than the long linker sequence of 20 amino acids. The affinity of CD22BBζ CAR-scFv with a short-linker sequence (6.10 nM) is not significantly different from that of CD22BBζ CAR-scFv with a long-linker length (1.70 nM) [59, 60]. The affinity of scFv can be measured with the equilibrium dissociation constant (KD) toward monovalent TAA in solution. The KD value and affinity of CAR-scFv of mAb are inversely related. The affinity of CAR-scFv is often considered an important parameter for evaluating the specificity and sensitivity of TAA. The affinity of scFv of mouse mAbs ranges from 1 micromolar (μM) to 1 nanomolar (nM). To date, no guidelines are available for developing optimal CAR affinity for a given antigen. However, relevant retrospective studies evaluating clinical and preclinical data on the affinity of CAR for solid cancer cells have shown that an intermediate affinity (KD of ~20–100 nM) of CAR-scFv has better clinical efficiency than a higher affinity [50, 61]. However, this finding does not apply to the Food and Drug Administration (FDA)-approved 19CAR binding domain. The scFv of FMC63 mAb has been used in three prototypic 19CAR-T cells and has high affinity (KD of 0.33 nM) [57, 58]. A novel CD19 CAR-scFv of CAT mAb has 42-fold lower affinity (KD of 14 nM) than that of CAR-scFv of FMC63 mAb. Although both share a similar binding epitope, the relatively lower affinity of CAR-scFv of CAT mAb results in lower activation-induced cell death (AICD), CD19 antigen trogocytosis, or antigen reduction after CD19 binding. Moreover, persistence of the low-affinity of 19CAR-scFv of CAT mAb were superior compared to high affinity 19CAR-T cells in pediatric patients with r/r B-ALL [62, 63]. The low-affinity of CAR-scFv of CAT mAb decreases severe off-target toxicity [64]. In contrast to monovalent affinity, polyvalent interaction between multiple receptors, particularly dimerized CAR and ligands, increases the overall binding strength (functional avidity) and thus antigen sensitivity. For example, CAR dimerization by two cysteine residues of the proximal region of the CD8α transmembrane domain (TMD) increases the functional avidity of CAR-T cells. Moreover, the CD8α TMD of CAR-T cells stabilizes the surface expression of CAR in T cells [65, 66]. The extracellular location of the TAA, and its accessibility for binding CAR-T cells is determined by the hinge region of the CAR ectodomain. Post-translational modification by N-linked glycosylation of the CD28 hinge domain facilitates CAR surface expression [67]. Moreover, CAR using the CD28 hinge forms a homodimer through its cysteine residue. Furthermore, CD28 TMD of CAR facilitates the formation of a heterodimer with the endogenous CD28 molecule of T-cells through the conserved YxxxxT motif [68]. Axicabtagene ciloleucel contains a CD28 hinge, CD28 TMD, and CD28 co-stimulatory domain, and thus has higher TAA sensitivity than FDA-approved lisocabtagene maraleucel (Liso-cel) and tisagenlecleucel (Tis-cel) ( Figure 1 ) [52, 69–71]. The immunoglobulin (Ig) like domain of the CD8α hinge and TMD (~5 nm) of Tis-cel; Ig like domain of CD28 hinge and TMD (~6 nm) of axicabtagene ciloleucel (axi-cel); and mutant IgG4-based long hinge and CD8α TMD (~7 nm) of Liso-cel show a non-classical immune synapse structure after ligand binding [12, 52, 68, 71, 72]. The spatial boundary of the immune synapse is approximately 15 nm. According to the segregation model of T cell activation, use of a proper hinge size (<15 nm) of CAR-T cells efficiently excludes the negative regulator of phosphatase CD45 from the non-classical immune synapse before antigen-dependent phosphorylation signals are transmitted by kinases [73, 74]. Although the CD8α hinge resists hinge proteolysis, modification of the hinge length (39 amino acids) decreases the treatment-associated adverse effects of Tis-cel with 19BBζ (71) CAR-T cells. Notably, the long CD8α hinge length of 55 aa in 19BBζ (86) CAR-T cells is associated with a diminished incidence of severe CRS and ICAN in patients with r/r B-NHL, but promising clinical efficacy [69, 75].

2.2 Costimulatory domains for CAR
Incorporation of the costimulatory domain 4-1BB (BB) results in more than 1000-fold in vivo expansion and 10-year long-term persistence of CD4+19BBζ CAR-T cells in patients with chronic lymphocytic leukemia (CLL) [76, 77]. In contrast, first-generation 19ζ CAR-T cells lacking the co-stimulatory domain have an in vivo persistence of 7 days in patients with LBCL, and human anti-mouse CAR-scFv has been reported; this treatment does not produce potent anti-tumor effects [78–81]. After activation, the cytoplasmic domain of 4-1BB constitutively potentiates non-canonical NF-κB signaling, thereby downregulating Bim apoptotic proteins, up-regulating anti-apoptotic B cell lymphoma-extra-large (Bcl-xL), and activating ERK of 19BBζ CAR-T cells [82]. Moreover, uptake of 4-1BB in CAR promotes mitochondrial biogenesis, mitochondrial fatty acid oxidation, and metabolic reprogramming of central memory CAR-T cells. Faster in vivo expansion of CD8+19CAR-Tcm cells than CD4+19CAR-Tcm cells has been observed [83, 84]. In addition, antigen independent CAR-scFv self-aggregation and tonic 4-1BBCD3ζ signaling continually activate TRAF2-NF-κB signaling and Fas dependent cell death [85]. Toxicity to T cells can be mitigated by attenuating CAR expression through the self-inactivating non-long terminal repeat (LTR) promoter of a lentiviral vector [86]. Moreover, 4-1BB co-stimulation ameliorates the exhaustion of 19BBζ CAR-T cells compared with 1928ζ CAR-T cells [58]. Furthermore, 19BBζ CAR-T cells ameliorate exhaustion by recruiting THEMIS-SHP1 phosphatase, which decreases the phosphorylation of CAR-CD3ζ [87]. Moreover, 19BBζ CAR-T cells have slower tumor rejection kinetics than 1928ζ CAR-T cells after initial antigen binding [88]. Intracellular signaling strength balances the effector function and memory development of CAR-T cells. Incorporation of the CD28 tyrosine residue YMNM-PYAP” proximal to, and CD3ζ distal to, the cell membrane facilitates binding of the p85 subunit of phosphatidylinositol 3-kinase (PI3K) and IL-2 secretion [89]. Activation of the PI3K-Akt-mTOR-PKC-theta-NF-κB pathway regulates glucose metabolism and T cell differentiation. In addition, adaptor proteins such as IL-2-inducible T-cell tyrosine kinase (Itk), filamin A, Lck, and Grb2/Vav interact with the distal carboxy-terminal proline-rich PYAP motif of the co-stimulatory domain of CD28. The involvement of the Slp-76/linker for activated T cell (LAT) proteins is less than that in native TCR signaling [90, 91]. Several downstream effectors lead to transcriptional activation of the canonical activated protein 1 (AP1) c-Jun, and Ca2+ flux-mediated calcineurin-nuclear factor of activated T cells (NFAT) pathway [92]. Thus, 1928ζ CAR-T cells with effector memory (EM) exhibit greater amounts of phosphorylated proteins and cytotoxic molecules, and faster in vivo tumor rejection kinetics than 19BBζ CAR-T cells during antigen recognition [93]. Percentage of EM phenotype in the final products of 1928ζ CAR-T cells are demonstrated to be one of key factors that positively affect complete clinical response in r/r DLBCL patients [94]. Phospho-proteomic analysis of activated 1928ζ CAR-T cells revealed a large abundant intracellular phosphorylation signals, which resulted in activation-induced cell death [87, 95, 96], because the in vivo proliferation kinetics and long-term persistence of CAR-T cells are closely correlated with the duration of clinical remission [97]. Genetic methods have been developed to balance the effector or memory function of 1928ζ CAR-T cells. For example, engineering of 1928ζITAM CAR-T cells to reserve an ITAM of CD3ζ proximal to the T cell membrane prolongs the in vivo persistence of 1928ζITAM CAR-T cells. In contrast, the intensity of intracellular signaling and antigen sensitivity is significantly attenuated [98]. In addition, substitution of CD3ζ in 1928ζ CAR-T cells with a CD3ɛ ITAM and CD3ɛ signal sequence decreases signal intensity and cytokine production through recruitment of the inhibitory Csk kinase [99]. Furthermore, genetic mutation of asparagine to phenylalanine in YMNM in CD28 decreases terminal differentiation of EM cells and exhaustion of 28ζ CAR-T cells [100].
Third-generation CAR-T cells have been developed by selecting and positioning one or more co-stimulatory domains from members of the CD28/B7 IgG superfamily, and/or members of the tumor necrosis factor receptor type II (TNFR II) family, such as OX40, CD27, and CD40 [101]. Recently, an alternative type III transmembrane protein, B cell-activating factor receptor (BAFF-R), has been identified as a new potent costimulatory domain of CAR-T cells [102]. Direct fusion of CD28 and the 4-1BB costimulatory domain of 1928BBζ CAR-T cells has more favorable in vivo persistence in patients with minimal r/r NHL after autologous HSCT than 1928ζ CAR-T cells [103, 104]. Similarly, direct fusion of inducible T-cell co-stimulator (ICOS) proximally, and 4-1BB distally, from the cell membrane prolongs the survival and in vivo persistence of 19ICOSBBζ CAR-T cells [105]. In contrast to the direct infusion of two co-stimulatory domains, use of parallel CAR constructs for co-expression of membrane proximally positioned CD28 and 4-1BB results in functional persistence and enhanced anti-tumor rejection [106]. Moreover, parallel 1928ζ CAR constructs co-expression of membrane proximal 4-1BB ligand (L) not only support 1928ζ-41BBL persistence but also activate interferon regulatory factor 7 dependent type I interferon production. The release of IFNß further activates innate and adaptive anti-tumor immunity [107]. In another well studied case, parallel 20BBζ CAR incorporation of full-length antigen independent OX40 into T cells recruits TNR associated factors 2 and 5, thereby activating the NF-κB pathway, the anti-apoptotic gene BCL-2, and the PI3K-AKT pathway. The combination of 20BBζ CAR signaling and antigen independent full-length OX40 costimulatory signaling reduce 20BBζ-OX40 CAR-T cells apoptosis, exhaustion, and improved in vivo persistence, anti-lymphoma efficacy compared with 20BBζ CAR-T cells [108].
A novel multiple chain chimeric immunoreceptor construct composed of ITAM with activating protein of 12 Da (Dap12) covalently binding the transmembrane domain of killer immunoglobulin-like receptor (KIR)S2, and KIRS2 conjugated to an antigen-specific scFv have been investigated. Notably, the antigen-specific KIRS2/Dap12 has stable surface expression and potent anti-tumor activity. After ligation of the antigen, KIRS2/Dap12 binds Syk and Zap70 kinase [109]. Optimization of CD19-scFv-KIRS2/Dap12-4-1BB engineered T cells by adding the co-stimulatory domain 4-1BB has been found to result in 100% CR and optimal IL-2 secretion, faster proliferation and persistence, and potent anti-tumor activity, as compared with non-co-stimulated CD19-scFv-KIRS2/Dap12. In addition, no ICANs and CRS have been detected in four adult patients with r/r B-ALL [110].
2.3 CAR delivery with viral vectors
To date, most autologous CAR-T cells and universal allogeneic CAR-T cells are produced ex vivo. The human immunodeficiency-derived third-generation self-inactivation lentiviral vector (SIN) with the U3 region of the 3´ long terminal repeat (LTR) removed minimizes the risk of replication-competent recombination. SIN LTR lentiviral vector constructs containing a central polypurine Tract (cPPT) sequence increase the transduction rate [111]. However, integration of CAR by lentiviral vectors at high multiplicity of infection results in multiple copies of CAR per T-cell genome and genotoxicity of the viral vectors. The FDA recommends that the viral vector copy number per CAR-T cell be fewer than five copies [112]. However, maintaining constitutive CAR transcription and protein expression with fewer than five copies per CAR-T cell is challenging. The selection of robust internal enhancer and promoter sequences can overcome epigenetic silencing, in addition to the introduction of a Woodchuck hepatitis virus post-transcriptional regulatory element in the 3´ untranslated region of the lentiviral vector of SIN LTR, the incorporation of a chromatin domain insulator flanking the properly sized transcription unit, and the addition of scaffold/matrix attachment regions [113–116]. The murine stem cell virus-based γ-retroviral vector is used to make axi-cel, and the murine stem cell virus LTR controls the transcription of axi-cel. Intron-containing promoters such as Elongation Factor 1α are used to drive transcription of CAR in lentiviral vectors. Despite the efficiency of viral vector delivery [117], the immunological barrier of the cytosolic DNA sensor to the viral vector sequence, the epitope encoded by the viral vector, the immune response to the transgene, and the transcriptional repression of the viral vector remain challenges [118–121]. In addition, the distribution of the lentiviral vector (LV) integration sites influences clinical treatment outcome by modulating in vivo proliferation of 19CAR-T cells in patients with chronic lymphocytic leukemia (CLL). Specifically, integration into the host T-cell methylcytosine dioxygenase TET2 allele together with a hypomorphic mutation in the second TET2 allele resulted in TET2 gene mutation. A single clonal proliferation of TET2-disrupted 19CAR-T cells with a less differentiated central memory phenotype induced CLL remission [119, 122, 123].
2.4 Precision genome engineering of CAR
Site-specific integration of CAR into the host genome has demonstrated clinical safety and efficacy. This method relies on the introduction of a site-specific break in the genome and the presence of homologous DNA sequences at double-stand breaks to perform high-fidelity homologous recombinational repair in host cells. In mammalian cells S or G2 phase, site-specific DNA double-stand breaks (DSB) could be generated by nucleofection of clustered regularly interspaced short palindromic repeats (CRISPR) associated system (Cas)9 and specific CRISPR Cas9 single guide (sg) RNAs efficiently and relatively precise. CRISPR Cas9/sgRNA is derived from the type II CRISPR/Cas system of microbial nuclease system. Moreover, rational design of the length and sequences of homologous arm of donor template DNA is key to HRR-mediated genome editing. Methods of delivering homologous donor template DNA include chemical transfection, pseudovirus particles from biological methods, and physical methods. Ex vivo electroporation, recombinant adeno associated virus (rAAV) donor vectors, non-integrating lentiviral vectors are efficient in transferring transgene incorporating donor template DNA [124, 125]. Another non-viral transgene approach for site-specific insertion of transgenes is the fusion of a highly soluble Sleeping Beauty (SB) 100X transposase with a catalytically dead Cas9 (dCas9). The choice of integration sites of SB 100X-dCas9 depends on the design of the gene-specific sgRNA. SB 100X transposon gene insertion sites are located approximately 300 base pairs downstream of sgRNA targets [126, 127]. Specifically, non-viral PD-1 locus integrated CD19-4-1BB-CD3ζ CAR-T cells in eight patients with r/r B-NHL have resulted in an 87.5% CR rate and durable responses for 1 year without grade 3 toxicity. This treatment has outperformed LV-19BBζ CAR-T cells [128]. Although genetic PD1 gene deletion reverses immunosuppression by PD-1 ligands [129], long-lasting PD-1 mRNA knockdown by stable transgenic expression of PD-1 targeted short hairpin (sh) RNA decreases surface PD-1 on CD19 CAR-T cells and impairs long-term anti-tumor function, particularly at lower ratios of effector cells to target cells. Similarly, an anti-PD1 blocking mAb accelerates CAR-T cell early differentiation and maturation, attenuates proliferation, and diminishes survival [130, 131]. Different from targeting CAR to the PD-1 locus, CC chemokine receptor 5 (CCR5) locus, adeno-associated virus site 1 (AAVS1) locus, and ß2M locus, targeting CAR to the TCRα chain constant region (TRAC) locus disrupted endogenous TCR, enhanced T cell potency after engineering. Specifically, integration of CAR to the TRAC locus averts 1928ζ CAR-T cells tonic signaling, delays 1928ζ CAR-T cells differentiation and exhaustion through optimal regulation of surface CAR levels after repetitive antigen stimulation. Surface 1928ζ CAR at the TRAC locus are downregulated within 12 hours (h) of antigen binding. The density of surface 1928ζ CAR in T cells gradually increased at 24 h after initial CD19 antigen binding. Recyclable 1928ζ CAR at the TRAC locus through the endocytosis pathway favored less-exhausted TRAC-1928ζ CAR-T cells after repeated antigen exposure [128, 132]. Data demonstrated that CD8+CAR-T cell function is abrogated following endogenous TCR activation. The TRAC site-specific CAR integration minimizes competitive activation of endogenous TCR signaling on CAR-T cell [133]. Binding of cognate ligands initiates ubiquitination of lysine residues of the cytoplasmic domain of CAR and lysosomal degradation of polyubiquitinated CAR [134, 135]. To ameliorate the loss of surface CAR in LV integrated CAR-T cells, a recyclable CAR that lacks the lysine residues of the intracellular domain can block the CAR ubiquitination pathway and promote CAR recycling to the T cell surface. TRAC locus integrated CAR-T cells have a dynamic uniform CAR surface density, thereby bypassing the CAR ubiquitination pathway [136].
2.5 Manufacturing CAR-T cells with younger phenotypes
In addition to patient status, age, tumor burden, and tumorigenic mutations, the numbers and quality of CD3+T cells in peripheral blood during leukapheresis in an individual patient may serve as clinical biomarkers for predicting the anti-tumor efficacy of autologous CAR-T cell products [35, 137].
Collection of CD3+T cells from adult cancer patients receiving T cell-impairing drugs, who have chronic infections and rapidly progressive disease, poses a challenge to providing good leukapheresis products before CAR-T cells production. In addition, thymus involution occurs with age and is accompanied by decreasing naïve T cell egress from thymus [138]. Clinical data have shown that a CD3+ T cell count above a threshold of 553/μL is associated with rapidly proliferating expansion kinetics in the peripheral blood 7 days after tisagenlecleucel infusion, and favorable progression free survival (PFS) and OS in patients with r/r DLBCL [139, 140]. Moreover, an increased frequency of naïve or stem-cell memory phenotype (CD3+Tscm) of CD8 T cells apheresis is independently associated with a sustained response to tisagenlecleucel [141, 142]. In contrast, senescence CD8+CD57hiCD39hiCD28low 19CAR-T cells or exhausted CD8+PD-1hiLAG3hi 19CAR-T cells are closely associated with decreased in vivo proliferative capacity and cytotoxicity in non-responders with r/r DLBCL [143]. For the treatment of adult patients with r/r LBCL, the dosage selection of axicabtagene ciloleucel, tisagenlecleucel, and Liso-cel should consider the available numbers of transduced 19CAR in CD3+ T cells of autologous final CAR-T cells product, patients’ body weight (kg), tumor burden, CAR constructs, the choice of pre-conditioning lymphodepletion regimen [144–146]. Strategies are needed to increase the efficacy and achieve the necessary amounts of CAR-T cells by supplementation with targeted drugs during the CAR-T cell production process. The production of autologous CAR-T cells involves activation of CD3+ T cells, CAR delivery by viral or non-viral methods, and an expansion process to achieve the required numbers. Preparing cellular starting material before CAR-T cell manufacture, enrichment of less-differentiated Tscm cells in leukapheresis peripheral blood mononuclear cells is ideal for overcoming T cells functional exhaustion of cancer patients. However, T cells derived from most adult patients with r/r DLBCL have no active metabolic status or terminal differentiation, and may be exhausted and express detectable immune checkpoint molecules. Moreover, in vitro activation of CD3+ T cells and CAR transduction with lentiviral vectors lead to T cell differentiation and exhaustion. Numerous developments have been made to shorten the in vitro manufacturing time and achieve transient cessation of CAR-T cell activation through the selection of epigenetic or enzymatic inhibitors to produce less differentiated “young” CAR-T cells [147, 148]. T cell exhaustion is a process of epigenic modification characterized by the expression of several key transcription factors, such as TOX and Nuclear Receptor Subfamily 4 Group A Member 3 (NRA4). NRA4 suppresses T cell factor 1 (TCF1). NFAT of activated T cells also activates the TOX/NRA4 axis [149–153]. In addition, Enhancer of Zeste 2 (EZH2), the catalytic subunit of the repressive Polycomb complex 2, silences memory-associated genes through trimethylation of histone 3 lysine 27 [154]. In addition, DNA methyltransferase 1 (DNMT1), DNA methyltransferase 3B (DNMT3B), and DNA methyltransferase 3A (DNMT3A) induce genome-wide de novo DNA methylation in terminally exhausted T cells [155, 156]. Recently, studies have focused on enrichment of CAR-Tscm cells or active metabolic CAR-T cells by modifying CAR-T cell transduction or expansion processes with small molecule drug inhibitors targeting epigenetic or metabolic enzymes. For example, addition of 10 nM decitabine, an inhibitor of DNA methyltransferase, synchronously with tandem CD19/CD20 CAR transduction, enriches human CAR-T cells with a memory phenotype [157]. In another case, the FDA-approved Src kinase inhibitor dasatinib targeting Lck or Fyn has been found to attenuate proximal CAR-T cell signaling and to be associated with less differentiated CAR-T cells during ex vivo expansion [148]. Similarly, TWS119, an agonist of the Wnt/β-catenin pathway, arrests effector T cell differentiation and enriches CD8+ stem-cell memory T cells (Tscm) [158]. As a cytokine supplement, the γc cytokine family member IL-15 downregulates mTOR activity and increases enzymes associated with fatty acid oxidation, thereby maintaining the metabolic state of the Tscm population [159, 160]. IL-21 acts synergistically with IL-15 during lentiviral transduction and expansion, thereby enriching functional CD8+CAR-Tcm cells [161, 162]. Moreover, overexpression of the canonical AP-1 of heterodimeric c-Jun/fos prevents exhaustion of CAR-T cells [163].
2.6 Target cell killing mechanism of CAR-T cells
Understanding how TAA specific CAR-T cells eliminate cancer cells could help develop better T cell engineering strategies. Ligand binding-induced CAR-scFv aggregation forms a non-classical immune synapse between CAR-T cells and tumors. The disorganized pattern of Lck, the multiple interactions between lymphocyte function-associated antigen (LFA)-1 and intercellular adhesion molecule 1 (ICAM-1), and the mechanical force at the non-classical immune synapse may mediate the effector functions of CAR-T cells, such as the secretion of cytokines and cytolytic enzymes into targeted cancer cells. The kinase Lck mediates abundant CD3ζ phosphorylation, and stimulates faster and higher basal activation of CD28CD3ζ CAR-T cells. THEMIS-SHP1 counteracts the extent of CD3ζ phosphorylation and mild activation of 4-1BBCD3ζ CAR-T cells in non-classical immune synapses [87]. Granzymes initiate the caspase-dependent mitochondrial apoptosis pathway and caspase-independent permeabilization of the mitochondrial outer membrane. In contrast, disruption of the nonclassical immune synapse-dependent release of perforin and the granzyme pathway is one mode of resistance to CAR-T cell therapy. The interaction of CD58−/− r/r DLBCL with CD2+19CAR-T cells is lost, thereby weakening the formation of non-classical immune synapses with cancer cells, and helping cancer cells escape killing by 19CAR-T cells [164, 165]. Similarly, in IFNγR1−/− glioblastoma, ICAM is downregulated at the transcriptional level. ICAMlo IFNγR1-/- glioblastoma failed to form a non-classical immune synapse with LFA1+ CAR-T cells, and thus glioblastoma escaped CAR-T cell killing [166]. Moreover, the Fas ligand homotrimer of activated CD8+CAR-T cells mediates the trimerization of the Fas (death) receptor in cancer cells and leads to apoptosis. Similarly, Fc-receptor-activated CAR-T cells of the extracellular spacer domain of IgG CH2CH3 lead to AICD in vivo through the Fas ligand-Fas receptor axis. Chronic TAA exposure and repeated stimulation induce AICD of CAR-T cells [71, 167]. In contrast, cancer cells with mutagenesis of Fas associated death domain (FADD), BH3-interacting-domain death agonist (BID), caspase 8, and TNF receptor superfamily member 10B (death receptor 5) escape apoptosis in the presence of CAR-T cells [168, 169]. In particular, NOXA, a protein of the Bcl2 family, has been identified as a central regulator of resistance to CAR-T cells in r/r DLBCL. NOXA is potential biomarker for r/r DLBCL non-response [170]. Moreover, TNF-associated apoptosis-inducing ligands (TRAIL and CD253) selectively trigger apoptosis of tumor cells expressing TRAIL death receptors (DR). For example, TRAIL-R1 (DR4) specific CAR-T cells have shown cross cancer antitumor efficacy in preclinical studies. The combination of TRAIL-R2 (DR5) CAR-T cells targeting myeloid-derived suppressor cell (MDSC) and MUC1-specific CAR-T cells against breast cancer have shown superior anti-tumor potential against breast tumors [171, 172]. Moreover, caspase 3-mediated gasdermin E-dependent pyroptosis in B-cell cancers is associated with cytokine release syndrome [21, 173] ( Table 1 ).

3. Challenges and solutions of autologous second-generation CD19 specific CAR-T cells therapy in r/r DLBCL
Second-generation CD19-specific CAR-T cells promised a good clinical outcome in approximately 43% of treated r/r DLBCL patients within one year. However, to increase the clinical response rate for those non-responders, T-cell engineering strategies have been developed. To overcome low TAA density tumors, immunosuppressive TME and CAR-T cell exhaustion, the development of bispecific tandem or loop CAR recognition domains, human leukocyte antigen-independent synthetic TCRαβ double-chain receptors integrated into the constant region of the TCRα chain, and armored CAR-T cells were presented in the following sections ( Figure 2 ).
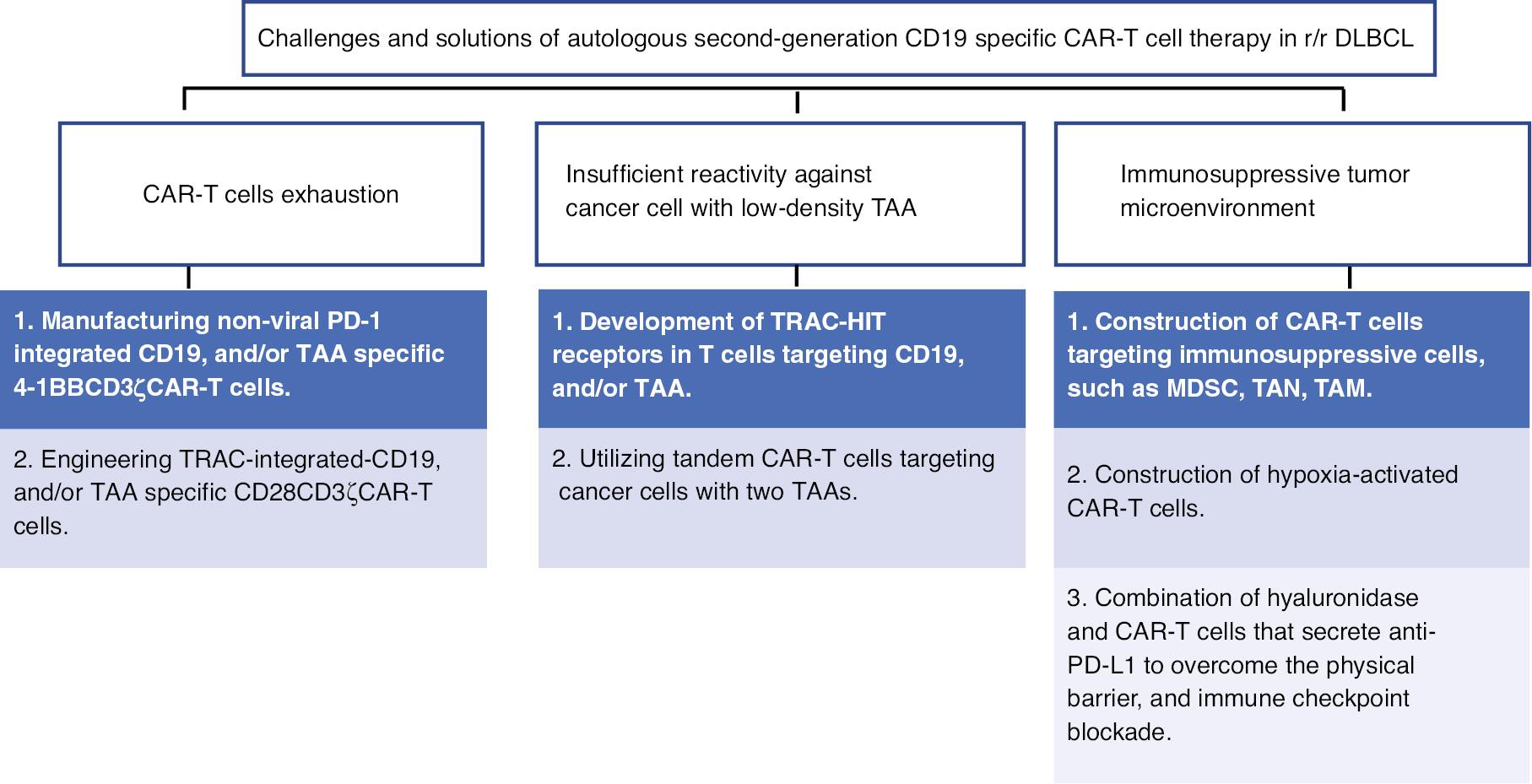
Challenges and solutions in CAR-T cell therapy for r/r DLBCL.
The illustrated chart lists demonstrate three types of challenges in the current CAR-T cell therapy for the treatment of patients with r/r DLBCL. The proposed solutions are indicated. MDSC, myeloid derived suppressor cell; TAN, tumor associated neutrophils; TAM, tumor associated macrophages; TRAC, TCRα chain constant region; HIT, human leukocyte antigen independent TCR αβ receptor.
3.1 Bispecific CAR-T cells
Dual antigen specific CAR-T cells have efficacy and clinical safety for patients with CD19+ and/or CD19−/low relapse, or native B cell cancers co-expressing two TAAs [174, 175]. Bispecific CAR-T cells have been clinically applied in several strategies, including the simultaneous transduction of two separate CAR vectors into T cells or the use of a bicistronic CAR construct for expression of two separate CARs in a single vector, the parallel-production of two separate antigen-specific CAR-T cells, administered sequentially within a day (cocktail CAR-T cells), or the sequential infusion of two antigen-specific CAR-T cells within a short time interval. Notably, after undetectable CD19BBζ CAR-T cells in peripheral blood of the patient, sequential administration of humanized CD22BBζ CAR-T cells within short time intervals promotes expansion of initial shrinking CD19BBζ CAR-T cells in the r/r B-NHL patients [176]. Moreover, a cocktail of CD22-28BBζ CAR-T cells and CD19-28BBζ CAR-T cells has been found to result in an ORR of 72.2% and 50% CR after treatment of 38 patients with r/r B-NHL. The median PFS was 9.9 months, and the median OS was 18.0 months after a median follow-up of 14.4 months [177]. In addition, the combination of high-dose chemotherapy, autologous stem cell transplantation, and a cocktail of CD22-28BBζ CAR-T cells and CD19-28BBζ CAR-T cells has been found to achieve a CR of 83.3% in 42 patients with aggressive r/r DLBCL after a median follow-up of 24.3 months [178].
In the development of a dual scFv in a single CAR design, product development of bispecific CAR-T cells could be evaluated more consistently. Bispecific tandem CARs have been developed by linking two separate scFvs in tandem. Bispecific loop CARs have been developed by linking two separate scFvs in a loop structure. The advantages of bispecific tandem or loop CAR is functional bivalency when bind to two separate TAAs of a target cell. By increasing the overall avidity of the non-classical immune synapse between the bispecific CAR-T cells and cancer targets, the “on-target” effects are increased, and the “off-target” effects are deceased. Notably, tandem CD20 (leu-16 mAb)/CD19 (FMC63 mAb) BBζ CAR-T cells have shown superior clinical safety and high efficacy in treating patients with r/r DLBCL: a 79% ORR, 71% CR, and 64% 12-month PFS have been achieved in 33 patients with r/r B-NHL in a phase I/II clinical trial. In long-term follow-up, an impressive PFS of 27.7 months was observed. Importantly, tandem CD20/CD19 BBζ CAR-T cells are associated with diminished ICAN severity [175, 179–181]. Bispecific tandem CD19 (FMC63 mAb)/CD22 (humanized) BBζ CAR-T cells have been found to achieve an ORR of 87.5% and a CR of 62.5% in the treatment of 16 patients with r/r DLBCL. The 2 year OS and PFS rates were 77.3% and 40.2%, respectively, in the phase I clinical trial. Of note, 16 patients with r/r B-NHL had no ICAN and little occurrence of severe CRS [182]. Bispecific looped CD19 (FMC63 mAb), VH-CD22 (M971 mAb), and VL-CD22VH-CD19VL BBζ CAR-T cells have achieved a 62% ORR and 29% CR in 21 patients with r/r LBCL ( Table 2 ) [183].
Comparative advantages and disadvantages of different forms of CAR-T cells targeting two tumor-associated antigens.
| Forms of CAR-T cells targeting two tumor-associated antigens | Advantages | Disadvantages |
|---|---|---|
| Sequential administration or CAR-T cell cocktails by mixing of two separate antigen-specific CAR-T cells | ||
| Generation of dual CAR-T cells through simultaneous transduction of a T cell with two different CAR vectors, thus resulting in two scFvs in a single T cell | ||
| Bispecific tandem CAR-T cells with two distinct scFvs connected in tandem in a single CAR construct | ||
| Bicistronic CAR vectors encoding two different CARs on the same T cell |
3.2 Native TCRαβ receptor and endogenous CD3 signaling machinery
Thymus-derived mature TCRαβ are highly sensitively to human leukocyte antigen (HLA)-loaded exogenous intracellular peptides (pep/HLA) and are approximately 100-fold more sensitive than 1928ζ CAR-T cells. Experimental data have indicated that approximately 1–50 ligands of the non-self-peptide/HLA complex per target cell triggers CD8+CTL activation signals. In addition, one to four exogenous Pep/HLA complexes per target cell triggers CD4+T cell activation. However, the presence of at least 3000 CD19 molecules per target cell triggers 1928ζ CAR intracellular signal transduction in T cells. The CD19 density of a normal B cell is ~20,000 molecules per cell. Low CD19 density (clone SJ25C1) is more common in CD10-positive DLBCL than CD10-negative nodal DLBCL samples [184]. A CD19 site-density of 952 molecules per r/r DLBCL biopsy has been found to result in CD19low relapse and progression 3 months after initial diagnostic CR [183]. Follicular lymphoma have CD19low antigen in 79% cases [185]. The decrease in CD19 antigen may be mediated by impaired membrane trafficking [186]. Trogocytosis of 19CAR-T cells has resulted in a decrease in CD19 in malignant B cells in a co-culture experiment [187]. A low CD22 density of less than 3000 molecules per malignant B cell impairs the functionality and persistence of 22BBζ CAR-T cells [59]. Furthermore, the CD20 site density is ~100,000 molecules per normal mature B cell [184]. CD20 expression below the level of 25,000 molecules per B-cell lymphoma is regarded as a low CD20 expression, and might not benefit from rituximab-based chemoimmunotherapy [188]. Native TCRαβ have a KD of 1–100 μmol/L toward foreign peptide-loaded HLA, whereas CAR-T cells have a KD of approximately 0.001 μmol/L toward the native protein epitope. Direct comparison of antigen sensitivity between CAR-T and native TCRαβ is difficult, owing to different antigen recognition models and intracellular signal activation by antigen binding [189, 190].
TCRαβ is a non-covalent heterodimer non-covalently associated with three preferred subunit pairs of CD3γɛ, CD3δɛ, and CD3ζζ. The TCRαβ-CD3 octamer has a stoichiometry 1:1:1:1 of TCRαβ:CD3γɛ:CD3δɛ:CD3ζζ dimeric modules. Each CD3γ, CD3δ, and CD3ɛ chain has a single ITAM, and the CD3ζ chain has three tandem ITAMs. At the molecular level, thymus-derived somatic V-diverse-(D)-junction (J) recombination, and the addition and removal of nucleotides at the VD and DJ junctions in complementary determining region 3 (CDR3) of the TCR variable (V)β chain of a developing T cell clone are responsible for the immune surveillance of various exogenous Pep/HLA complexes. CDR3 of TCR-Vα is determined by V and J, as well as by the addition and removal of nucleotides at the VJ junctions of developing T cell clones [191]. CDR3 of both TCR-Vα and TCR-Vβ recognizes the specific Pep/HLA complex. CDR1 and CDR2 of both TCR-Vα and Vβ contact the side chains of HLA alleles [192]. In addition, CD4 or CD8 coreceptors are costimulatory receptors that enhance the interaction between TCRαβ and pep/HLA. The involvement of the CD8β coreceptor enhances the TCRαβ response against low-affinity Pep/HLA ligand during thymic positive selection, through the formation of a TCRαβ-coreceptor zipper [193]. In addition, eight conserved amino acid peptides of the membrane-proximal region of the TCR-α chain constant region (TCR-Cα-CPM) promote close approach to the CD8 coreceptor and antigen reactivity [194, 195]. Importantly, a homologous genetic mutation in the TCR-α constant chain (TRAC) impairs assembly and/or intracellular transport of the TCR-αβ complex, thus resulting in a lack of surface expression of the TCR-αβ complex and of thymic egress of TCR-αβ cells in patients [196].
The presence of an immunological synapse composed of TCRαβ and pMHC complexes facilitates a cascade of proximal transmembrane signaling. The mature immunological synapse consists of the peripheral supramolecular activation cluster, as well as the central supramolecular activation cluster, which is enriched in TCRαβ, coreceptors, costimulatory receptors, inhibitory receptors, phosphorylated tyrosine kinase Lck, Src homology region 2 (SH2)-containing ζ-chain-associated protein kinase 70 (ZAP70), cytoplasmic protein tyrosine phosphatases (PTPs) of SH2-containing protein tyrosine phosphatase 2 (Shp2), and PKC-theta [197, 198]. Several molecular models have been proposed to explain how TCR-αβ binding of the non-self-peptide HLA triggers signal transduction of CD3 complex consisting of three dimers of CD3γɛ, CD3δɛ, CD3ζζ. According to the conformational change model, Pep/HLA binding triggers the movement of the proximal C-terminus of the CD3ζ transmembrane helix and the exposure of ITAMs of CD3ζ [199]. The Src-family kinases p56Lck and p59FYN phosphorylate ITAMs (YXXL/V) of CD3ζζ, CD3γɛ, and CD3δɛ. Phosphorylated ITAMs recruit several key enzymes and adaptors including Lck kinase, ZAP70, and phosphorylated adaptor of LAT, thus further amplifying the activation signal [199]. Phosphorylated LAT acts as a docking site that recruits multiple signaling proteins associated with the T-cell membrane (LAT signalosomes) and results in activation of several downstream pathways [200]. The recruitment of Lck to the intracellular tail of CD4 or CD8 coreceptors, and the cyclic interaction of CD4 or CD8 with the β2-domain of HLA-DR or the α3-domain of HLA-A2 amplify the T-cell response to foreign antigens by a million-fold [201]. In addition, CD3ζ K33 polyubiquitination mediated by the E3 ligase Cbi-b and Itch counteracts the phosphorylation of CD3ζ of activated T cells via the endocytic pathway [134]. CARs are designed to be independent of endogenous TCR/CD3 signaling. Activated 1928ζ CAR-T cells have fewer phosphorylated linkers for activated T cells (LAT) in comparison with canonical TCR signaling. LAT phosphorylation is not detected in activated 19BBζ CAR-T cells [96, 202]. Increasing evidence indicates that enhancement of intracellular signaling strength after antigen recognition increases the antigen sensitivity of CAR-T cells [96, 203]. Low density tumor antigen sensitivity of 19BBζ CAR-T cells is enhanced by activation of the phosphorylated LAT-signaling pathway through the insertion of the CD3ɛ signal sequence [96].
3.3 Human leukocyte antigen-independent TRAC locus integrated synthetic TCRαβ
To eliminate tumor cells with low-density antigens, an ultra-sensitive human leukocyte antigen independent synthetic TCRαβ receptor (HIT) has been constructed by replacing the endogenous TCR-Vβ and TCR-Vα with VH and VL of scFv, respectively. The synthetic chimeric VHCβ and VLCα heterodimer was integrated and expressed in the TRAC locus (TRAC-HIT) of recipient T cells by CRISPR/Cas9/sgRNA and rAAV donor template. Immunoglobulin VH and VL of TRAC-HIT directs surface antigens recognition of target cell, and CD3γɛ, CD3δɛ, CD3ζζ three dimers-mediated signaling in TRAC-HIT T cells. Moreover, the TRAC-HIT-receptors provide higher antigen sensitivity than that of TRAC-28ζ CAR-T cells. Experimental data show that as few as 20 surface CD19 molecules per B-cell cancer can be recognized and killed by TRAC-HIT-receptors modified T cells after phosphorylation of extracellular signal regulated kinase (pERK), CD3ζ, CD3ζ associated protein kinase (ZAP70), and degranulation. In contrast, TRAC-28ζ CAR-T cells can kill target cells carrying 200 CD19 molecules. Similarly, low abundance target of approximal 370 surface CD70 molecules per acute myeloid leukemia (AML) could be efficiently eliminated by CD70-knockout CD70 specific TRAC-HIT T cells. Moreover, constitutive expression of the CD80/4-1BB ligand in CD70-knockout CD70 specific TRAC-HIT T cells significantly improved its in vivo survival and anti-AML potency compared with CD70-knockout CD70 specific TRAC-CAR-T cells [204].
In contrast to the construction of the TRAC-HIT receptor, the mutant synthetic T-cell receptor and antigen receptor (mutSTAT) has been constructed by grafting the endogenous TCR-Vβ and TCR-Vα with VL and VH of scFv, and replacing the human TCR-Cβ and TCR-Cα sequences with corresponding murine sequences. Furthermore, an additional disulfide bond was introduced between the chains in the murine (m) TCR-Cβ(S57C) and mTCR-Cα (S48C), and a hydrophobic sequence was introduced into the transmembrane domain of mTCR-Cα (S48C) to improve surface display and achieve proper pairing between VH-mTCR-Cα (S48C) and VL-mTCR-CαCβ (S57C) chains. In addition, the mTCR-Cα-connecting peptide of mutSTAT function in correctly pairing of TCR/CD3 complex after T cell engineering. Lentiviral vector-mediated transduction of mutSTAT is effective against solid cancers, particularly solid cancer cells with low antigen density, which outperform 28ζ CAR-T cells. In the resting state, mutSTAT has lower tonic signaling than 28ζ CAR-T cells [205]. Moreover, mutSTAT co-stimulated with OX40 significantly prolongs the in vivo persistence. CD19 specific mutSTAR-OX40 has been found to have a 100% CR rate in treating 18 patients with r/r B-ALL 4 weeks post infusion in a phase I clinical trial: 75% (12/16) patients remained leukemia-free after a median follow-up of 545 (433–665) days; 55.6% patients (10/18) had mild CRS; and two patients had grade III neurotoxicity [206]. In another preclinical study, VL and VH domain of the Fab of human anti-CD19 (ET190L1, clone) were fused with the δ chain and γ chain of TCR respectively to direct CD19 antigen specificity and antigen dependent CD3γɛ/CD3δɛ/CD3ζζ signaling, cytokine production, degranulation. Significantly, Fab antibody TCRγδ engineered T cells had less differentiated and exhausted Tscm phenotype, and a lower CRS than 19CAR-T cells in patients derived xenograft leukemia model [207]. In another construct design, a TCR fusion construct (TRuCs) contained an anti-CD19 scFv bound to one of the full-length TCR/CD3 octamer subunits, including TCR-Vα, TCR-Vβ, CD3γ, CD3δ, or CD3ɛ, via a flexible glycine serine linker. TRuCs became an integral component of the TCR-CD3 octamer and did not affect the Pep/HLA-mediated T cell response. Although TRuC-T and CAR-T differ in the quality of intracellular signaling, TRuC-T elicits a potent anti-tumor response in both hematologic and solid cancer xenograft mouse models [208].
3.4 Targeting the immunosuppressive tumor microenvironment
Immune checkpoints, tumor stroma, immunosuppressive cells, hypoxia, and metabolic limitations in the TME of r/r DLBCL hinder CAR-T cell therapy [209]. Infiltration of the TME with immunosuppressive CD4+T cells, exhausted CD8+T cells, myeloid cells, normal B cells, cancer-associated fibroblasts (CAFs), chronic hepatitis B viral infection, and potential cell-cell interaction between malignant B cells and tumor-infiltrating cells promote tumor cell survival and immune invasion [210, 211]. Moreover, activation of hypoxia-inducible factor 1 alpha in r/r DLBCL upregulates the enzyme hexokinase 2, thus promoting glucose metabolism and facilitating tumor growth under hypoxic stress. However, under hypoxic stress (<2% O2 concentrations), expansion, cytokine production, and granzyme B release of second-generation CAR-T cells are markedly diminished. Decreases in protein synthesis and defective CAR-T cell metabolism in TME have been reported [212–214]. Hypoxia-activated CAR-T cells have been engineered to adapt and survive in the TME [215]. CAFs promote r/r DLBCL survival, angiogenesis, and cancer niche formation. CAFs upregulate the expression of fibroblast activation protein (FAP), smooth muscle α-actin (αSMA), programmed cell death ligand 1 (PD-L1), and PD-L2, thereby suppressing PD1+CAR-T cells [216]. Patients with r/r DLBCL expressing PD-L1 have shown poor overall survival [217]. Hyaluronidase and checkpoint blockers against anti-PD-L1 manipulated CAR-T cells degrade hyaluronic acid in the extracellular matrix, and facilitate deep CAR-T cell invasion in the TME [218]. In addition, resident endothelial cells and fibroblastic reticular cells upregulate PD-L1, thus transforming lymph node podoplanin+ fibroblastic reticular cells into CAF-like immunosuppressive cells. Armored CAR-T cells secreting PD-1 blocking scFv have been engineered to overcome PD-L1 mediated immune suppression [219]. Data have indicated that infiltration of tumor associated macrophages from r/r DLBCL TME is negatively associated with remission status after 19CAR-T cell therapy [220]. In addition, CD11b+HLA-DR− myeloid derived suppressor cells are associated with poor PFS and OS of 19CAR-T cells [221]. Metastasis of B cell lymphoma is associated with the epithelial-to-mesenchymal transition process [222]. Hyaluronan synthase 2 (HAS2), and tumor growth factor β (TGFβ) regulate epithelial-to-mesenchymal transition [223] via phosphorylation of SMAD1/5 [224]. Overcoming the tumor suppressor TGFβ by constructing both TGFβ- and TAA-responsive CAR-T cells may expand the therapeutic window of r/r DLBCL [225, 226]. In addition, limited trafficking and infiltration of CAR-T cells into the TME are challenges, particularly for patients with extranodal lesions of r/r DLBCL in soft tissue [227, 228]. Modification of CAR-T cells expressing specific chemokines directs CAR-T cells into the TME. Local injection of 19CAR-T cells in combination with intravenous infusion and radiotherapy before 19CAR-T cell therapy are effective alternative methods [229, 230]. PETN-deficient r/r DLBCL are infiltrated with a large numbers of myeloid derived suppressor cells (MDSC), Tregs, and abundant expression of indoleamine 2,3-dioxygenase (IDO1) protein, an immunosuppressive enzyme involved in a rate-limiting step of tryptophan catabolism. PETN-deficient r/r DLBCL exhibited reduced infiltration of CD8+CTL and NK cells compared with suppressive immune cells, which establish immunosuppressive TME in r/r DLBCL after PETN-deficiency [231]. To activate the immune response, peptide vaccines, oncolytic virus, and Toll like receptor agonists have been developed to recruit T cells into the TME in r/r DLBCL. A peptide cocktail vaccine consisting of a 21 amino acid peptide from IDO (DTLLKALLEIASCLEKALQVF) and a 19 amino acid peptide from PD-L1 (FMTYWHLLNAFTVTVPKDL), together with nivolumab (anti-PD-1), has achieved an ORR of 80% and 43% CR with a median PFS of 26 months. CD4+ T cells and CD8+ T cells have been found to infiltrate into tumor sites in responder patients [232].
4. CONCLUSION
CD19 specific CAR-T cells do not induce clinical remission in approximately 57% of patients with relapsed and/or refractory DLBCL. In tumors with TAA heterogeneity especially low density surface expression of TAA, the immunosuppressive tumor microenvironment poses challenges for the current CAR-T cell therapies. Moreover, CAR-T cell exhaustion under an immunosuppressive tumor microenvironment TME potentially limit its long-term efficacy. With the rapid development of CAR design, and novel T cell engineering strategies, evidences have accumulated that a synthetic T cell receptor, independent of human leukocyte antigen, delay engineered T cell exhaustion, and have at least 10 fold super TAA sensitivity than CD28 co-stimulated CARs. Moreover, the development of bispecific tandem CAR-T cells or loop CAR-T cells not only maximizes clinical safety but also overcomes the heterogeneity of TAAs of cancer cell. Future autologous and/or allogeneic T cell engineering technologies are gradually being developed to target the dynamic, complicated TME and identify combinatory targets against the TME to address unmet clinical needs.