
Discovery and Development of Diabetic Cardiomyopathy
With economic growth and improved living standards, the incidence of diabetes has significantly increased, and the age of onset has gradually decreased, thus extending the overall course of diabetes. Throughout the prolonged course of diabetic cardiomyopathy (DCM), the risk of cardiovascular disease markedly increases, because of glucose and lipid metabolism disorders. The concept of DCM was first introduced by Rubler et al. in 1972, who autopsied patients with heart failure with diabetic glomerulosclerosis and found no other clear causes of heart failure beyond diabetes [ 1]. The currently accepted definition of DCM includes a definitive history of diabetes accompanied by clear structural and functional changes in the myocardium, excluding ischemic diseases, hypertension, and other potential causes of myocardial changes. The progression of this disease includes a silent, asymptomatic subclinical stage, typically of long duration, with initial myocardial structural damage leading to diastolic dysfunction, followed by systolic dysfunction, and eventually late-stage heart failure [ 2].
Substantial numbers of patients have diabetes, most of whom are in a prolonged subclinical stage of DCM without overt cardiac symptoms. Although cardiomyopathy in prediabetes shares several mechanisms with diabetes, such as hyperglycemia-induced damage, oxidative stress, and inflammation, it also includes unique factors associated with the transitional state of glucose intolerance characteristic of prediabetes, such as insulin resistance, lipotoxicity, and microvascular dysfunction. These mechanisms suggest that cardiomyopathy can begin developing even before diabetes becomes clinically evident; therefore early detection and management of prediabetes are important in preventing cardiomyopathy and other cardiovascular diseases. Cardiomyopathy can even begin to develop before the clinical manifestations of diabetes become apparent. Echocardiography, a non-invasive method crucial for assessing cardiac structure and function, is particularly important for identifying subclinical left ventricular dysfunction. Echocardiographic indices such as the myocardial performance index and presystolic wave are closely associated with left ventricular dysfunction in type 2 diabetes and serve as valuable indicators [ 3, 4]. Cardiac magnetic resonance imaging provides detailed images of cardiac structure and function, thereby enabling precise assessment of myocardial texture, ventricular volume, and myocardial activity, and capturing disease progression. Other assessments such as measurement of serum biomarkers (including brain natriuretic peptide (BNP) and its N-terminal prohormone (NT-proBNP), and advanced glycation end-products (AGEs)) and functional cardiac tests can also be used to evaluate cardiac function, particularly in subclinical stages [ 5].
Mitochondrial Dysfunction and Diabetic Cardiomyopathy
Mitochondria, as the energy supply centers of cells, play critical roles in maintaining overall metabolic balance. Mitochondrial dysfunction can lead to severe metabolic imbalances and various disease states [ 6]. A large proportion of patients with type 2 diabetes exhibit insulin resistance, accompanied by elevated blood glucose and insulin levels. The toxic effects of high glucose and insulin levels, combined with energy production disorders caused by insulin resistance, affect several processes, including mitochondrial substrate utilization, oxidative stress, post-translational modifications, mitochondrial dynamics, and mitochondrial calcium uptake ( Figure 1). These disruptions collectively contribute to mitochondrial dysfunction [ 7]. Addressing mitochondrial dysfunction through pharmacological or other interventions has increasingly become a focus of research aimed at developing new strategies for treating DCM.
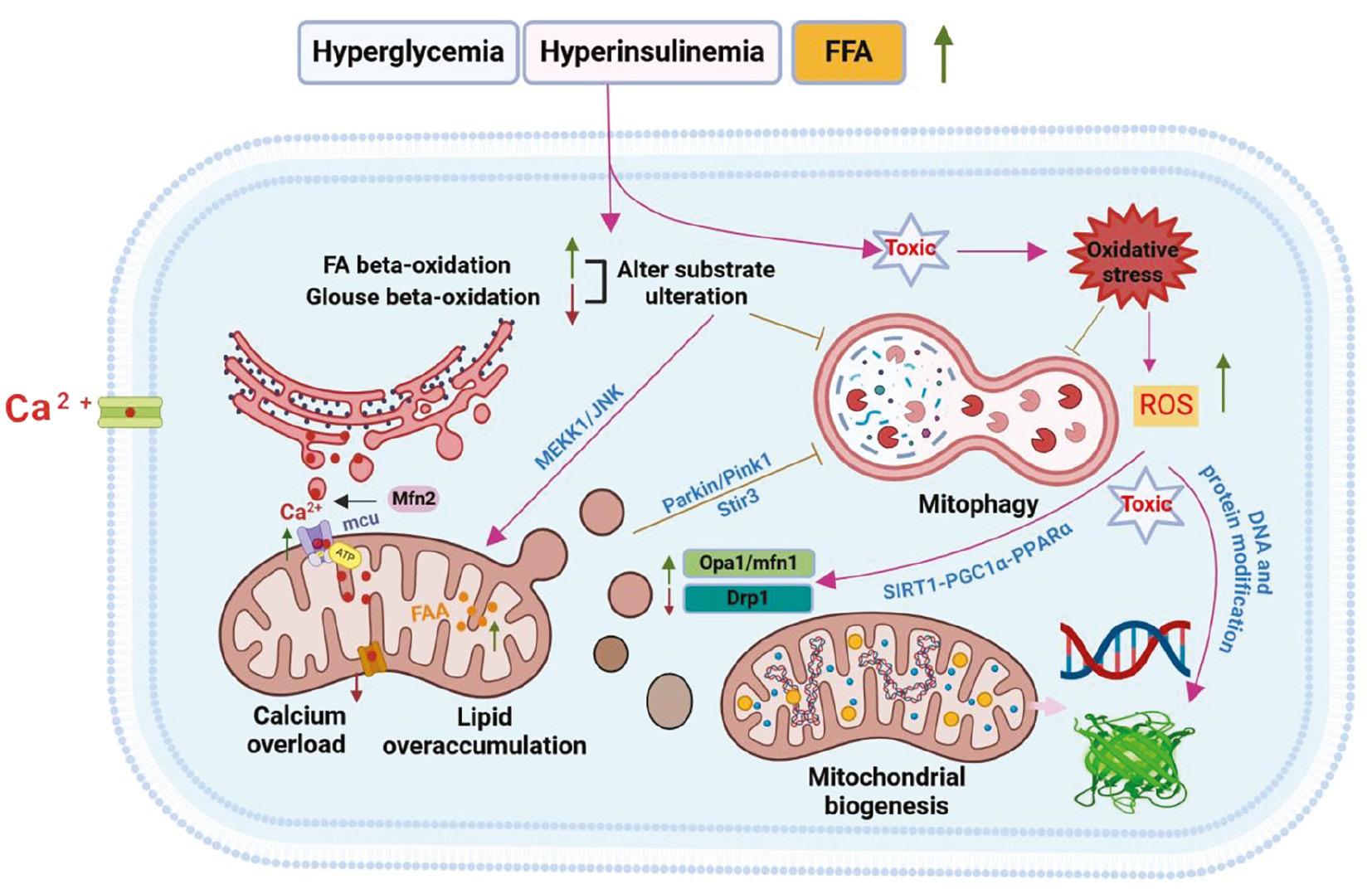
Perturbed Calcium Ion Homeostasis Regulation, Dysregulated Mitophagy And Mitochondrial Biogenesis, Impaired Mitochondrial Energetics, DNA and Protein Modification, As Well As Altered Substrate Utilization in the Pathogenesis of DCM.
FA: fatty acid; ROS: reactive oxygen species; Mfn2: mitochondrial fusion protein 2; MEKK1/JNK: mitogen-activated protein kinase 1/c-Jun N-terminal kinase; Pink1: PTEN-induced kinase 1; SIRT3: Sirtuin-3; SIRT1-PGC-1α-PPARα: Sirtuin-1-peroxisome proliferator-activated receptor coactivator-1 α-peroxisome proliferator-activated receptor-α; Drp1/Mfn1: Dynamin-related protein 1/mitochondrial fusion protein 2; Opa1: optic atrophy 1.
New Therapeutic Targets and Drugs for Ameliorating Diabetic Cardiomyopathy in Mitochondrial Dysfunction Interventions
Calcium Regulation Disorder
Mitochondria-related calcium regulation disorder is a crucial mechanism in the development of DCM. Mitochondria play major roles in energy metabolism and calcium ion regulation in myocardial cells. Under diabetic conditions, mitochondrial calcium channel impairment can result in imbalances in calcium ion influx and efflux. These imbalances increase mitochondrial internal calcium ion concentration, and subsequently lead to mitochondrial dysfunction and cellular damage. Given that the mitochondria are crucial sites of intracellular energy production, mitochondrial functional status is closely associated with redox states [ 8]; therefore, intracellular calcium ion homeostasis is also associated with redox states. Numerous studies have indicated that high glucose levels in diabetic states lead to mitochondrial dysfunction [ 9] and abnormalities in calcium ion channels, thereby resulting in excessive intracellular calcium ion accumulation, calcium overload, and increased reactive oxygen species (ROS) production, and creating a vicious cycle of mitochondrial dysfunction and apoptosis. Mitochondrial dysfunction often begins with the opening of mitochondrial permeability transition pores (mPTPs), which in turn alters mitochondrial membrane permeability, decreases mitochondrial membrane potential, and leads to irregular calcium influx into the cytoplasm causing calcium overload [ 6]. Calcium modulators, such as trimetazidine, ranolazine, nifedipine, and verapamil, are widely used in clinical settings, primarily for treating angina, arrhythmias, heart failure, and hypertension. Most of these drugs act by directly targeting calcium channels, such as nifedipine, whereas others, such as trimetazidine and ranolazine, exert their effects by regulating the energy metabolism of ion pumps or through Na(+)/Ca2(+) exchange.
In recent years, numerous substances and targets have been found to effectively ameliorate calcium regulation disorders in DCM. Natural compounds have broad effects. Ferulic acid, a commonly found phenolic compound and major component of cell walls, has been shown by Palayyan et al. to ameliorate pathological changes caused by high glucose, such as decreased mitochondrial membrane potential, calcium overload, and oxidative stress, through the SERCA/PLN (sarcoplasmic reticulum Ca(2+)-ATPase/phospholamban) pathway; therefore, this compound might potentially be a valuable health supplement [ 10]. Liu et al. have discovered that rhynchophylline mitigates intracellular calcium overload and protects the mitochondrial structure by downregulating phosphorylation of the ryanodine receptor [ 11].
The transport of Ca(2+) involves many key structures whose regulation can ameliorate calcium ion regulation disorders. The mitochondrial calcium uniporter (MCU), which regulates mitochondrial Ca(2+) uptake, is crucial for maintaining free mitochondrial calcium concentration. Julieta et al. have found that cardiomyocytes exposed to simulated high glucose conditions show reduced Ca(2+) and MCU protein levels. Overexpression of MCU restores Ca(2+) in cardiomyocytes to normal levels, and consequently normalizes glucose oxidation and fatty acid oxidation [ 12]. The endoplasmic reticulum (ER) serves as an intracellular calcium reservoir, and coupling structures or transporters between the ER and mitochondria facilitate calcium transfer. Maya et al. have found that adenovirus-mediated overexpression of ER-mitochondria tethering proteins ameliorates mitochondrial calcium ion exchange disorder and diminished biological function; therefore, ER-mitochondria calcium uncoupling might play a reversible role in the early development of DCM [ 13]. Zhang et al. have found that mitochondrial fusion protein 2 (Mfn2) promotes ER-mitochondria contact in cardiomyocytes treated with high glucose. Silencing of Mfn2 maintains mitochondrial function, inhibits mitochondrial calcium overload, prevents mitochondrial apoptosis, and decreases ER stress, thereby increasing cardiomyocyte survival under high glucose conditions [ 14]. Brain and muscle ARNT-like protein 1 (Bmal1) is an essential transcription factor that regulates the circadian rhythm and participates in numerous cardiac disorders. Research by Zhang et al. has implicated Bmal1 in the development of DCM via B-cell lymphoma 2/inositol 1,4,5-trisphosphate receptor (Bcl2/IP3R)-mediated mitochondrial Ca(2+) overload; therefore, targeting the circadian clock (Bmal1) might be a potential treatment for DCM [ 15]. Sphingolipids are key mediators of cellular stress responses and effectors of mitochondrial function. Elevated levels of lactosylceramide, an initial product of glycosphingolipid formation, are associated with diminished mitochondrial respiration and calcium retention capacity in diabetic heart tissue. Sergei et al., in a type 1 diabetes model, have found that lactosylceramide effectively inhibits mitochondrial respiration and decreases calcium retention capacity; therefore, lactosylceramide is a primary sphingolipid associated with diabetic cardiac mitochondrial defects. The ceramide metabolism pathway is a promising target for correcting mitochondrial abnormalities associated with type 1 diabetes [ 16].
Mitochondrial calcium dysregulation in DCM is a complex issue involving calcium uptake, transport, and efflux. Understanding these specific mechanisms and developing new treatment methods to improve cardiac function in patients with diabetes is highly important. Recently discovered compounds such as FA and huperzine A have been shown to protect against mitochondrial calcium overload; however, their specific clinical translation will require further research. Currently, clinical trials are exploring the use of ranolazine and istaroxime, which have been shown to be effective treatments for end-stage heart failure. New research targets such as MCU, ER-mitochondria tethering proteins, Mfn2, and Bmal1, which are involved in calcium dysregulation in DCM, require further investigation to develop targeted drugs for clinical application.
Autophagy, Mitophagy, and Apoptosis
Autophagy is the process in which cells degrade damaged or aging organelles through the intracellular enzyme dissolution system [ 17]. Under physiological conditions, autophagy is an adaptive mechanism that allows cells to clear damaged mitochondria and maintain their own homeostasis, which is crucial for maintaining normal cardiac structure and function. Mitochondrial autophagy, also known as mitophagy, specifically involves the removal of unnecessary or damaged mitochondria. In diabetic states, high levels of glucose, lipids, and insulin damage mitochondria, thus leading to the accumulation of numerous damaged and functionally impaired mitochondria. Decreased mitochondrial autophagy hinders the clearance of damaged and disrupted mitochondria, exacerbates the cellular energy supply disorder, and eventually leads to cell apoptosis [ 18].
Recent studies have identified many drugs that ameliorate autophagy, mitophagy, and apoptosis in DCM cells. The mineralocorticoid receptor blocker eplerenone has demonstrated antifibrotic effects in diabetic hearts. Ramírez et al. have found that this treatment alleviates mitochondrial dysfunction and apoptosis without altering hyperglycemia [ 19]. Exposure of cardiomyocytes to high glucose significantly increases ROS generation, decreases mitochondrial membrane potential (ΔΨm), leads to release of cytochrome c, and increases Bcl-2 and BCL2 Associated X, Apoptosis Regulator (Bax) expression [ 20]. Potent liver X receptor (LXR) agonists such as T0901317 (T0), Astragalus polysaccharides, fibroblast growth factor 21 (FGF21), and dihydromyricetin effectively decrease apoptosis, lower ROS levels, and ameliorate mitochondrial dysfunction [ 21– 24]. Studies have shown that mitochondrial aldehyde dehydrogenase (ALDH2) and helix B surface peptide (HBSP) might protect against diabetes-induced cardiac dysfunction by improving attenuated autophagy in diabetic cardiomyocytes in an AMPK-dependent manner. These drugs therefore warrant further investigation [ 25, 26]. Hydrogen sulfide (H 2S) is the third endogenous gas confirmed to have cardioprotective effects. Recent research has indicated that H 2S lowers oxidative stress; prevents mitochondrial damage; activates autophagy through the AMPK/mTOR pathway; and decreases mitochondrial apoptosis by regulating Mfn2, a key protein linking mitochondria and the ER. Another study has found that H 2S promotes mitophagy by increasing the S-sulfhydration of Ubiquitin Specific Peptidase 8 (USP8), thereby enhancing Parkin deubiquitination by recruiting Parkin to mitochondria [ 27– 29].
Several meaningful targets have also been discovered. Lin28a, a highly conserved RNA-binding protein first reported in Caenorhabditis elegans in 1984, is involved in the regulation of cell proliferation and pluripotency. Sun et al. have shown that Lin28a knockdown exacerbates diabetic damage, and leads to a decreased left ventricular ejection fraction (LVEF), increased apoptosis index, and worsened mitochondrial cristae destruction. Overexpression of Lin28a significantly improves LVEF in diabetic mice, promotes autophagy, decreases myocardial apoptosis index, and alleviates mitochondrial cristae damage, and therefore might have therapeutic value. Mitochondrial calcium uptake 1 (MICU1), a key regulator of mitochondrial Ca(2+) uptake, plays important roles in regulating mitochondrial oxidative phosphorylation and redox balance. A recent study has shown that reconstituting MICU1 increases mitochondrial Ca(2+) uptake, and subsequently activates the antioxidant system, and inhibits high glucose and high fat-induced cardiomyocyte apoptosis [ 30]. Increasing the expression or activity of MICU1 might serve as a pharmacological approach to ameliorating DCM.
Numerous studies have investigated specific targets and drugs acting on mitophagy. For instance, Yu et al. have found that in cardiomyocytes of DCM mice, the expression of SIRT3, a soluble protein in the mitochondrial matrix, is associated with cardiac dysfunction, interstitial fibrosis, cardiomyocyte apoptosis, and mitochondrial damage, accompanied by inhibited autophagy and mitophagy. Whereas SIRT3 knockout exacerbates these effects, SIRT3 overexpression in vitro activates autophagy and mitophagy, thereby decreasing mitochondrial damage and cardiomyocyte apoptosis [ 31]. Additionally, Zhang et al. have found that treatment of diabetic mice with polyamines enhances cardiac function and autophagy, in a manner mediated by upregulation of SIRT3 [ 32]. Melatonin, an indole heterocyclic hormone secreted by the pineal gland, has been shown by Ding et al. to rescue impaired mitophagy activity in DCM, potentially through inhibition of macrophage stimulating 1 (Mst1) and activation of Parkin translocation [ 33]. Recent studies have shown that the selective bromodomain inhibitor JQ1 inhibits Bromodomain-containing protein 4 (BRD4), a member of the BET family of epigenetic regulatory genes, and improves mitophagy. Resveratrol regulates mitophagy through the AMPK/SIRT1-mediated Inositol-requiring enzyme 1α (IRE1α)/PINK signaling pathway, and leads to delayed fibrosis in DCM [ 34, 35]. Ginseng Dingzhi decoction, a traditional Chinese medicine, decreases heart failure and protects cardiomyocytes. Wang et al. have found that this medicine regulates mitophagy and mitochondrial homeostasis, and its combination with metformin enhances mitophagy levels and protects cardiomyocytes [ 36].
Research on mitochondrial autophagy remains in preliminary stages, and many studies have focused on the key proteins regulating mitochondrial autophagy. Drugs such as aripiprazole, melatonin, and FGF21 are already widely used in clinical treatments, and H 2S has been demonstrated to have therapeutic effects on various diseases, although its safety and clinical translation require further evaluation. Sirt3 is a notable mitochondrial autophagy target being investigated in many ongoing studies. The role of mitochondrial autophagy in DCM is complex, and further in-depth research will be necessary to reveal its specific mechanisms and potential treatment strategies.
Mitochondrial-Related Modifications
Mitochondrial-related modifications in DCM refer to a series of changes in the structure, function, and metabolic pathways of mitochondria under diabetic conditions. These modifications include morphological changes, protein post-translational modifications, and regulation of mitochondrial gene expression. As the main site of nutrient metabolism, mitochondria produce intermediate metabolites, such as lactate and succinyl-CoA, which mediate protein post-translational modifications. Lactylation (Kla), malonylation (Kma), β-hydroxybutyrylation (Kbhb), and succinylation (Ksucc) are derived from glucose metabolism, whereas crotonylation (Kcr), butyrylation (Kbu), propionylation (Kpr), glutarylation (Kglu), and 2-hydroxyisobutyrylation (Khib) are derived primarily from fatty acid oxidation and amino acid metabolism. Additionally, enzymes involved in mitochondrial metabolism are extensively regulated by modifications such as succinylation and crotonylation [ 37].
The close relationship between mitochondria and post-translational modifications, particularly novel acylation modifications, is crucial for understanding the principles of biological processes and disease mechanisms. O-GlcNAcylation is a post-translational modification occurring in multicellular organisms, targeting cytoplasmic, nuclear, and mitochondrial proteins. This process involves the glycosylation of proteins with N-acetylglucosamine (O-GlcNAc) associated with the hydroxyl groups of serine and threonine side chains in proteins. O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) are the modifying enzymes that control the dynamic balance of this protein modification in response to nutritional and stress signals [ 38– 40]. OGT uses UDP-GlcNAc to modify serine and/or threonine residues on various proteins. Hyperglycemia has been shown to increase mitochondrial O-GlcNAcylation. Banerjee et al. have analyzed the mitochondria of diabetic rat hearts and found elevated mitochondrial OGT and diminished OGA. OGT is located in the cytoplasm, whereas OGA is located in the mitochondria. The pyrimidine nucleotide carrier, a mitochondrial inner membrane transport protein, transports UDP-GlcNAc from the cytoplasm to the mitochondria. Knockdown of this transport protein significantly decreases mitochondrial O-GlcNAcylation. Inhibiting the activity of OGT or OGA in neonatal rat cardiomyocytes significantly affects energy production, mitochondrial membrane potential, and mitochondrial oxygen consumption. Therefore, cardiac mitochondria have a robust O-GlcNAc cycle, and dysregulation of O-GlcNAcylation might play a key role in diabetes-related mitochondrial dysfunction [ 41]. SIRT3 is a mitochondrial protein deacetylase that regulates mitochondrial function through post-translational modifications. Bagul et al. have found that resveratrol activates SIRT3, regulates the acetylation status of the mitochondrial transcription factor TFAM, and maintains mitochondrial function and cell size in diabetic rat hearts [ 42]. Fang et al. have also assessed whether resveratrol alleviates diabetic cardiac dysfunction by improving mitochondrial function through SIRT1-mediated peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) deacetylation. The study found improvements such as increased LVEF, fractional shortening, increased the ratio of early diastolic peak flow velocity to late diastolic peak flow velocity, and decreased expression of the hypertrophic markers ANP, BNP, and β-MHC [ 43]. Resveratrol is a potential drug for alleviating DCM, although its related mechanisms require further exploration. Adropin is a newly discovered bioactive protein encoded by energy balance genes that plays important roles in regulating energy metabolism and maintaining insulin sensitivity. Recent studies have indicated that adropin regulates the expression of mitochondrial acetyltransferase GCN5L1, and consequently alters the acetylation status and activity of metabolic enzymes, and promotes glucose utilization; these findings suggest potential future therapeutic approaches for DCM [ 44]. Ubiquitination, a post-translational modification, not only participates in protein degradation but also plays crucial roles in regulating cellular functions. Recent studies have indicated that lentinan (a β-1,3-glucan polysaccharide extracted from shiitake mushrooms) targets the caveolin-1 (CAV1)/succinate dehydrogenase subunit A (SDHA) pathway through ubiquitination, and consequently decreases SDHA degradation, and improves mitochondrial dysfunction and mitochondria-induced apoptosis [ 45].
Research on mitochondrial modifications has largely focused on post-translational modifications of mitochondrial proteins, importantly including the regulation of mitochondrial O-GlcNAcylation. Additionally, acetylation modifications regulated by Sirt3 have crucial regulatory functions, and resveratrol, which acts on Sirt3, is a potential drug for alleviating DCM. Studies on mitochondrial RNA and DNA modifications remain limited, and further increased investment is necessary.
Mitochondrial Oxidative Stress
Oxidative stress is a key factor in the onset and progression of DCM. Under normal conditions, a redox balance exists within cells, wherein the antioxidant system effectively removes free radicals and other oxidizing substances, and consequently protects cells against oxidative damage. However, in people with diabetes, owing to insufficient insulin secretion or insulin resistance, blood glucose levels rise and lead to a series of metabolic disorders and inflammatory responses. These factors disrupt the redox balance within cells and result in oxidative stress [ 2]. Mitochondria are important organelles for intracellular energy metabolism and are the main sites of ROS production [ 46]. In DCM, factors such as hyperglycemia and insulin resistance impair mitochondrial function and lead to mitochondrial oxidative stress. Mitochondrial oxidative stress can decrease mitochondrial membrane potential and ATP synthesis, and increase mitochondrial DNA damage, thus further exacerbating myocardial cell injury and dysfunction [ 47]. Moreover, mitochondrial oxidative stress promotes and worsens inflammatory responses, and can lead to myocardial cell death and fibrosis.
Currently, many studies are aimed at ameliorating mitochondrial oxidative stress to alleviate the onset and progression of diabetes. Mitochondria possess an intricate antioxidant enzyme system including superoxide dismutase, glutathione peroxidase, the thioredoxin system, and catalase [ 48]. Various drugs and targets have been identified to improve or enhance the activity of related antioxidant enzymes or inhibit oxidase activity, and consequently ameliorate oxidative stress. Early research by Baseler et al. has demonstrated that the overexpression of mitochondrial phospholipid hydroperoxide glutathione peroxidase 4 (mPHGPx) improves cardiac function in DCM mice [ 49]. Similarly, Guo et al. have found that the Rho-associated protein kinase (ROCK) inhibitor fasudil significantly decreases the abundance of mitochondrial Rho A, ROCK 1, and ROCK 2 proteins in DCM rats. Fasudil also restores the activity of myocardial mitochondrial succinate dehydrogenase (SDH) and monoamine oxidase (MAO); inhibits opening of the mPTPs; and decreases total antioxidant capacity, malondialdehyde, hydroxyl radicals, reduced glutathione, and superoxide dismutase levels in the myocardium [ 50]. Monji et al. have discovered that the glucagon-like peptide-1 receptor (GLP-1R) agonist exendin-4 (Ex-4), a drug used to treat type 2 diabetes, ameliorates myocardial mitochondrial oxidative stress by inhibiting NADPH oxidase 4 while increasing antioxidants (SOD-1 and glutathione peroxidase) [ 51].
Mitochondria are a major source of ROS in the heart, and therapeutic strategies targeting mitochondrial ROS may provide benefits in disease treatment. Ni et al. have investigated the mitochondrially targeted antioxidant mito-TEMPO, which decreases high glucose-induced intracellular oxidative stress levels and consequently ameliorates cardiac dysfunction. The protective effects of mito-TEMPO are associated with downregulation of ERK1/2 phosphorylation [ 52]. Similarly, Cao et al. have found that overexpression of mitochondrial aldehyde dehydrogenase 2 (ALDH2) decreases ROS production in high glucose-induced myocardial cell injury, thereby inhibiting the activation of nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome and pyroptosis [ 53]. Calcium-dependent protease 1 (Calpain-1), a member of the class of calcium-dependent proteases, is widely expressed in various organisms, and is associated with multiple physiological functions and pathological processes, such as focal cerebral ischemia, neurodegenerative diseases, and DCM. Recent research has shown elevated Calpain-1 protein and mitochondrial activity in the hearts of type 1 diabetic mice. This elevated mitochondrial calpain-1 is associated with increased mitochondrial ROS production and oxidative damage, as well as decreased ATP synthase-α (ATP5A1) protein and ATP synthase activity. Inhibiting Calpain or upregulating ATP5A1 has been found to increase ATP5A1 and ATP synthase activity, prevent mitochondrial ROS production and oxidative damage, and mitigate DCM changes in mice [ 54]. Recent studies have also found that in arrhythmogenic cardiomyopathy, the absence of apoptosis-inducing factor induced by Calpain-1 can lead to massive nuclear DNA fragmentation and cell death, and ultimately heart failure. Inhibition of Calpain-1 has also been found to alleviate the progression of heart failure in DCM [ 55]. N-acetylcysteine (NAC) exerts antioxidant and anti-inflammatory effects by increasing glutathione levels, thereby scavenging free radicals and decreasing oxidative stress. In vitro studies by He et al. have shown that NAC reduces palmitic acid-mediated mitochondrial ROS primarily through ER stress, and subsequently alleviates myocardial apoptosis, but has little effect on high glucose-induced cardiac injury [ 56]. Other studies have found that dietary docosahexaenoic acid (DHA) decreases mitochondrial oxidative stress in high fat-induced DCM [ 57]. Tom70, a mitochondrial outer membrane translocase, promotes the import of precursor proteins into mitochondria. Precursor proteins may be involved in the regulation of oxidative stress and mitochondrial function. Recent studies have shown that Tom70 overexpression in diabetic mice alleviates mitochondrial fragmentation and dysfunction, as well as oxidative stress in the hearts of db/db (leptin knockout) mice, thereby ameliorating cardiac hypertrophy, interstitial fibrosis, and ventricular dysfunction [ 58].
With ongoing developments of in materials science, new therapeutic methods are continually emerging. Zhang et al. have used PEGylated nanoliposomes combined with ultrasound-targeted microbubble destruction (UTMD) to effectively deliver nano acidic fibroblast growth factor (AFGF) to the heart tissue in diabetic rats. NM-AFGF inhibits diabetes-induced myocardial oxidative stress injury by activating the protein kinase B/Glycogen Synthase Kinase 3/Nuclear factor erythroid 2-related factor 2 (AKT/GSK/Nrf-2) signaling pathway, and ultimately improves myocardial structure and function in diabetic rats. This therapeutic strategy is therefore promising [ 59]. Recent studies have indicated that many natural compounds, including flavonoids, β-lapachone (Lap, a natural compound that enhances antioxidant activity in various tissues), 11-keto-β-boswellic acid (AKBA), dihydromyricetin (DHY), and irisin, have significant anti-mitochondrial oxidative stress effects [ 60– 65].
Non-coding RNAs (ncRNAs) are RNA molecules that do not encode proteins, but play important roles in gene expression regulation, cell differentiation, apoptosis, metabolism, and other physiological processes. Recent studies have shown that ncRNAs are also closely associated with DCM occurrence and development. The intronic long ncRNA Dachshund homologue 1 (lncDACH1) is closely associated with heart failure and cardiac regeneration. Recent studies on DCM have demonstrated that knocking out lncDACH1 decreases mitochondrial oxidative stress, apoptosis, myocardial fibrosis, and hypertrophy in DCM mice, and consequently improves heart function. Overexpression of lncDACH1 aggravates mitochondrial-derived ROS levels and apoptosis while decreasing manganese superoxide dismutase (Mn-SOD) activity. In contrast, silencing lncDACH1 decreases ROS production, mitochondrial dysfunction, and apoptosis in high glucose-treated cardiomyocytes, while increasing Mn-SOD activity. The mechanism might be associated with the ubiquitination-mediated degradation of SIRT3 in mouse hearts [ 66]. MicroRNAs (miRNAs) are endogenous small non-coding RNAs that play important roles in diabetes. Zhu et al. have found that db/db mice show significantly elevated expression of miR-340-5p in heart tissues and cardiomyocytes, thus leading to increased cardiomyocyte apoptosis, elevated ROS production, and impaired mitochondrial function. Inhibition of miR-340-5p with a tough decoy (TUD) vector has been found to prevent ROS production and apoptosis, and therefore might serve as a potential therapeutic target [ 67].
Oxidative stress and subsequent inflammatory damage are common pathological processes in visceral diseases. Mitochondria are the main organelles involved in energy production and are also the primary sites of ROS production. Some conventional antioxidants, such as N-acetylcysteine and glutathione have been widely used in clinical settings. Recently discovered substances such as fasudil, exendin-4 (Ex-4), mito-TEMPO, lncDACH1, and many traditional Chinese medicines will require further research to achieve clinical application and translation. Recent studies on ultrasound-targeted microbubble destruction technology, which delivers drugs specifically to diseased myocardium, is a highly promising research direction.
Mitochondrial DNA
Mitochondrial DNA (mtDNA), genetic material present within mitochondria, plays crucial roles in cellular energy metabolism and apoptosis. Recent studies have shown that mtDNA is closely associated with the occurrence and development of DCM [ 68– 70]. Hyperglycemia can lead to impaired mitochondrial function, thereby affecting mtDNA replication and repair, and can also promote oxidative stress, thereby leading to oxidative damage and an increased mutation rate in mtDNA. These factors can alter mtDNA copy number and mutation rates, and subsequently affect mitochondrial function and energy metabolism. Moreover, oxidative stress is a crucial factor affecting mtDNA function and energy metabolism. Oxidative stress can decrease mitochondrial membrane potential, reduce ATP synthesis, and increase mtDNA damage, thus further exacerbating myocardial cell damage and dysfunction. Finally, insulin resistance also affects mtDNA function and energy metabolism. Insulin resistance decreases glucose uptake in mitochondria and consequently affects mitochondrial energy metabolism. Additionally, it promotes oxidative stress, which in turn leads to oxidative damage and an increased mtDNA mutation rate.
Current research on mtDNA associated with DCM remains limited. Early studies by Hicks et al. have indicated that in rat cardiomyocytes under long-term high glucose treatment, the activation/inhibition state of mitochondrial topoisomerase changes and is associated with oxidative stress [ 71]. The Jagannathan team, through microarray technology combined with crosslinking immunoprecipitation and next-generation sequencing, has discovered that the mitochondrial microRNA pool redistributes in spatially different mitochondrial subgroups after injury. The redistributed mitomirs show significant interaction with the mitochondrial genome and regulate translation by inducing the production of associated silencing proteins, thus leading to pathological changes [ 72]. A common lesion in mtDNA damage is 8-hydroxy-2’-deoxyguanosine (8-OHdG). When 8-oxoguanine DNA glycosylase (Ogg1) fails to repair this lesion correctly, mutations can form. Cividini et al. have found that in the hearts of type 1 diabetic mice, Ogg1 protein levels are elevated, but Ogg1 activity is significantly lower than that in the hearts of control non-diabetic mice. The elevated levels of 8-OHdG and mtDNA damage observed in diabetic hearts suggest that hyperglycemia-induced Ogg1 O-GlcNAcylation increases mtDNA damage [ 73]. Recent studies have also indicated that certain substances ameliorate mtDNA damage. Mushtaq et al. have examined the expression of genes associated with mitochondrial signaling damage in diabetic rats and observed dysregulation in the expression of PGC-1α, cytochrome c, Mfn2, and Drp1. N-acetylcysteine, selenium, and ascorbic acid have been found to ameliorate mitochondrial dysfunction [ 74].
Research on the roles of mtDNA in DCM remains in initial stages. Inhibiting the formation of 8-OHdG in mtDNA holds promise as a new therapeutic target. Increased research efforts are hoped to provide new insights and methods for the treatment of DCM.
Mitochondrial Respiratory Chain Uncoupling
In DCM, mitochondrial uncoupling is a significant biological process involving loss of control over the mitochondrial inner membrane potential, thus leading to the uncoupling of the proton gradient between the inner and outer mitochondrial membranes (IMM and OMM, respectively), and decreasing the efficiency of ATP production. Consequently, mitochondria may produce more heat instead of ATP, while increasing the production of oxidative stress and cytotoxic substances, thereby accelerating myocardial cell functional damage. Mitochondrial uncoupling in patients with diabetes is typically associated with the following factors [ 75]: (1) Lipid metabolism abnormalities: In diabetic conditions, lipid metabolism abnormalities can lead to changes in mitochondrial membrane lipids and consequently affect the stability of the mitochondrial inner membrane potential. (2) Oxidative stress: Oxidative stress, a major feature of DCM, can cause oxidative damage to mitochondrial inner membrane proteins and subsequently affect membrane permeability. (3) Calcium ion imbalance: Calcium ions play essential roles in regulating mitochondrial function and metabolism. In DCM, calcium ion imbalance can disrupt the proton gradient between the IMM and OMM, and lead to uncoupling. (4) Inflammatory response: Inflammatory responses also play important roles in DCM. Inflammatory mediators can directly or indirectly affect mitochondrial structure and function, thereby promoting mitochondrial uncoupling. Mitochondrial uncoupling causes mitochondria to produce more heat, which is typically considered an adaptive response to oxidative stress. However, prolonged uncoupling can lead to energy metabolism imbalances and cellular damage that ultimately accelerate DCM progression. Therefore, blocking mitochondrial uncoupling may serve as a potential strategy for treating DCM.
Research on mitochondrial respiratory chain uncoupling in DCM remains limited. Earlier studies have found that male rats with diabetes induced by streptozotocin show diminished mitochondrial biogenesis pathways and assembly of electron transport chain (ETC) complexes targeting complex I, as well as oxidation of complex II and long-chain fatty acid substrates leading to electron leakage and superoxide production. Mitochondrial fatty acid oxidation and complex I defects are both causes of increased protein lysine acetylation. Quantitative analysis of mitochondrial lysine acetylation post-translational modifications has revealed 54 lysine acetylation sites on 22 mitochondrial proteins with higher acetylation levels in in patients with diabetes than controls. Therefore, mitochondrial protein lysine acetylation is a common consequence of increased fatty acid oxidation and complex I defects, which might contribute to the metabolic inflexibility of the diabetic heart [ 76]. Fortunately, many substances have been found to ameliorate mitochondrial respiratory chain dysfunction in DCM. Methylene blue (MB) is a compound reported to act as an alternative electron carrier in the mitochondrial ETC. Berthiaume et al. have found that administering MB at 10 mg/kg/day to diabetic rats promotes NADH oxidation, increased NAD(+) and activity of deacetylase SIRT3, and reduced protein lysine acetylation in the mitochondria of diabetic hearts. Further studies have revealed that MB treatment does not affect acyl-CoA dehydrogenase activity but decreases 3-hydroxyacyl-CoA dehydrogenase activity and long-chain fatty acid oxidation, thereby improving cardiac function [ 77]. Another study has found that administering MB (0.1 μmol/L) to DCM rats activates complex I and II substrates and increases mitochondrial oxygen consumption in both normal and diseased mitochondria. In the presence of complex I substrates (glutamate and malate), MB significantly increases H 2O 2 release but has opposite effects in mitochondria activated by complex II substrates (succinate). The calcium retention capacity of mitochondria did not differ between healthy and diabetic rats. These findings suggest that MB improves respiratory function in mitochondria isolated from the hearts of both diabetic and non-diabetic rats, and has a dual, substrate-dependent effect on ROS production [ 78]. Other studies have shown that the flavonoid dihydromyricetin enhances the activity of complexes I/II/III/IV/V and consequently improves mitochondrial function [ 24]. Uncoupling proteins (UCPs) are expressed in the myocardium and prevent free radical damage by regulating mitochondrial respiration, thereby decreasing ROS production. Studies have demonstrated that transgenic diabetic animals lacking UCPs are susceptible to oxidative damage and cardiac dysfunction [ 79]. A-kinase anchoring protein 121 (AKAP1), located on the OMM, plays a key role in regulating mitochondrial function. Qi et al. have observed downregulated AKAP1 expression in the hearts of STZ-induced diabetic mouse models. AKAP1-KO significantly worsens cardiac dysfunction in STZ-treated diabetic mice. Mechanistic explorations have revealed that AKAP1 deficiency inhibits complex I activity by preventing the translocation of the NADH-ubiquinone oxidoreductase 75 kDa subunit (NDUFS1) from the cytoplasm to the mitochondria. Restoration of AKAP1 expression promotes NDUFS1 translocation to the mitochondria. The study findings suggest that upregulation of AKAP1 has therapeutic potential for myocardial injury in patients with diabetes [ 80]. Vitamin B3, a precursor of NAD(+), exists in various forms, such as niacin and nicotinamide. These cofactors play crucial roles in cell homeostasis, mitochondrial respiration, ATP production, and ROS generation and inhibition. The niacin derivative (3-piperidin-2-hydroxy-1-propyl) nicotinic amidoxime (BGP-15) has been found to improve mitochondrial respiratory function in obese type II diabetic rats. The specific mechanism may be associated with the tricarboxylic acid (TCA) cycle and the ETC, although further elucidation is required [ 81].
Mitochondrial respiratory chain uncoupling is a major reason for diminished energy efficiency and the production of oxidative stress toxic products, often caused by disturbances in the ETC. The clinical detoxifying drug MB can serve as an alternative electron carrier compound in the mitochondrial ETC and has shown great potential for clinical application. Other vitamin-like drugs, such as vitamin B3 and the niacin derivative BP-15, also produce similar effects and have significant clinical value.
Mitochondrial Dynamics
To adapt to changing stress conditions, mitochondria undergo continuous fission and fusion to maintain the dynamic balance of the mitochondrial network, thus meeting energy metabolism and other biological needs. This process, known as mitochondrial dynamics, is crucial in regulating metabolism, inflammation, energy production, and cellular differentiation and movement, and provides a crucial foundation for maintaining cellular homeostasis. Recent studies have shown that mitochondrial dynamics is also closely associated with the occurrence and development of DCM. Specifically, in DCM, inhibition of mitochondrial dynamics leads to abnormal mitochondrial morphology, decreased function, and disrupted energy metabolism [ 82]. Studies have shown diminished rates of mitochondrial fusion in in patients with DCM, which might potentially lead to abnormal mitochondrial morphology and diminished function. In contrast, other studies have shown enhanced rates of mitochondrial fission in patients with DCM, which might decrease the number of mitochondria and disrupt energy metabolism.
Mitochondrial fission and fusion are regulated primarily by fission and fusion proteins, which play crucial roles in maintaining mitochondrial quality and function [ 83]. Mitochondrial fission is regulated primarily by Drp1, which is typically located in the cytoplasm. The activity of Drp1 is regulated by translocation as well as post-translational modifications, such as phosphorylation and ubiquitination. The main process of mitochondrial fission includes two steps: (1) when stimulation occurs, Drp1 is recruited to the OMM, where it interacts with other proteins involved in mitochondrial fission and forms a ring-like structure through oligomerization; (2) through GTPase activity, Drp1 hydrolyzes GTP, thereby changing the complex structure and causing the contraction of the IMM and OMM, and inducing mitochondrial fission. Mitochondrial fission factor (MFF) and mitochondrial fission protein 1 (Fis1) are Drp1 receptors that mediate the mitochondrial fission process. Additionally, mitochondrial dynamics proteins (MiDs) participate in the mitochondrial fission process. Mitochondrial fusion is the process through which individual mitochondria connect in a predetermined sequence under regulation by multiple proteins. This process includes mitochondrial trans-tethering, OMM fusion, and IMM fusion, and is regulated primarily by three proteins – OPA1, mitochondrial fusion protein 1 (Mfn1), and Mfn2 – through the OMM and IMM. OPA1 is involved primarily in the IMM fusion process, whereas Mfn1 and Mfn2 primarily regulate OMM fusion.
People with diabetes often have poor glucose control, thus leading to cardiomyocyte toxicity and affecting mitochondrial dynamics. High glucose levels in cardiac progenitor cells have been found to decrease cell viability and the expression of cell cycle-associated molecules (including CDK2 and cyclin E). Tube formation assays have demonstrated that high glucose significantly decreases the tube formation ability of cardiac progenitor cells. Fluorescence labeling of mitochondria in cardiac progenitor cells has demonstrated that high glucose alters mitochondrial dynamics and consequently increases the expression of fission-associated proteins, including Fis1 and Drp1. Additionally, specific blockade of GLUT1 has been reported to improve cell viability, tube formation, and regulation of mitochondrial dynamics in cardiac progenitor cells [ 84].
Moderate exercise has significant benefits in cardiovascular diseases. Veeranki et al. have found that moderate-intensity exercise significantly benefits cardiovascular health by restoring levels of connexins 43 and 37 (Cx43 and Cx37), and the mitochondrial biogenesis regulators Mfn2 and Drp1, thus preventing excessive mitochondrial fission and improving mitochondrial function. Similarly, Bækkerud et al. have found that intermittent exercise training partially restores mitochondrial fission and ameliorates mitochondrial dysfunction [ 85, 86]. SIRT1 is an important nuclear deacetylase widely involved in mitochondrial homeostasis and various biological processes such as apoptosis, inflammation, and metabolism. Certain substances have been found to be associated with its regulatory effects. Ma et al. have found that the SIRT1 activator resveratrol ameliorates impaired mitochondrial biogenesis. Additionally, melatonin and rosmarinic acid have been found to inhibit mitochondrial fission via the SIRT1-PGC1α pathway, directly downregulate Drp1 expression, and consequently alleviate diabetic cardiac dysfunction [ 33, 87, 88]. Other studies have indicated that the natural compound ophiopogonin D (OP-D) inhibits palmitic acid-induced mitochondrial fission and dysfunction, potentially by reversing the imbalance in DRP1 translocation, and decreasing expression of the fusion proteins MFN1/2 and OPA1 [ 89]. Decreasing endogenous apoptosis and inhibiting mitochondrial fission may be potential targets for delaying cardiac complications in patients with diabetes.
In recent years, new meaningful targets for improving mitochondrial dynamics have been discovered. Calcium sensing receptor (CaSR) is a G protein-coupled receptor involved in maintaining calcium homeostasis, and regulating cell proliferation and apoptosis. In DCM rats, significantly diminished CaSR expression, intracellular calcium levels, and cardiac function have been found to activate the gp78-ubiquitin proteasome system, induce myocardial energy metabolism disorders, and disrupt the structure and function of mitochondria and intercellular connections. Therefore, CaSR might be a potential target for treating DCM [ 90]. Mammalian sterile 20-like kinase 1 (MST1) is a homolog of yeast Ste20 kinase in mammals. As a core component of the Hippo inhibitory pathway, it plays important roles in regulating embryonic development, cell migration and differentiation, maintaining immune system stability, promoting apoptosis, and inhibiting tumor cell growth. Recent studies have demonstrated that MST1 gene knockout inhibits excessive mitochondrial fission, decreases left ventricular remodeling, and prevents DCM progression. This mechanism is associated with the upregulation of Drp1 expression; activation of Drp1(S616) phosphorylation and Drp1(S637) dephosphorylation; and promotion of Drp1 recruitment to mitochondria [ 91].
In some cases of DCM, mitochondrial fusion is inhibited. Recent studies have indicated that myocardial cells treated with the mitochondrial fusion promoter M1 under high glucose conditions show elevated OPA1 expression, thus promoting mitochondrial fusion, enhancing mitochondrial respiratory capacity, and decreasing the production of mitochondrially derived superoxides [ 92]. Recent studies have also discovered many substances that promote mitochondrial fusion and improve cardiac dysfunction in diabetes, such as arecoline (PG), melatonin, empagliflozin, and nicotinamide riboside. These substances promote OPA1-mediated mitochondrial fusion and ameliorate mitochondrial dysfunction through various pathways, including the PTP1B-Stat3, SIRT6-AMPK-PGC-1α-AKT axis, and SIRT1-PGC1 α-PPAR α [ 93– 96]. These findings suggest that promoting mitochondrial fusion might also be a potential strategy for treating DCM.
Mitochondrial biogenesis involves the transition between two mitochondrial states under the stimulation of complex external environments and stress: new mitochondria are produced through mitochondrial biogenesis, whereas damaged mitochondria are removed through mitophagy. Recently, many substances have been discovered that promote mitochondrial biogenesis under DCM conditions, such as salidroside, secreted frizzled-related protein 2 (SFRP2), D-β-hydroxybutyrate-(R)-1,3-butanediol monoester (ketone ester [KE]), tetrahydrobiopterin (BH4), and miR-144 [ 97– 101].
Mitochondrial dynamics involves mitochondrial fission, fusion, and biosynthesis. In diabetes, cardiomyocytes shoe disordered mitochondrial dynamics. Despite extensive research, contradictory conclusions have often been reported in this field. A synthesis of the current evidence suggests that promoting mitochondrial fusion and inhibiting excessive mitochondrial fission are therapeutically meaningful directions. Some widely studied drugs include melatonin, englitazone, and resveratrol. Mst1, OPA1, and Drp1 are also highly significant targets of action.
Mitochondrial Lipid Metabolism Disorders
In DCM, elevated levels of circulating fatty acids lead to abnormal accumulation in the heart. This accumulation causes lipotoxicity, exacerbates oxidative stress, and leads to cellular and organ dysfunction [ 102]. Under diabetic conditions, abnormal lipid metabolism in cardiomyocytes affects the function and structure of mitochondria. As the core organelles for energy production, the mitochondria experience energy production disorders, which exacerbate myocardial cell damage and worsen cardiac function. Changes in the substrates for mitochondrial energy production profoundly affect mitochondrial functional status. Under physiological conditions, cardiomyocytes use primarily fatty acids for energy, and, to a lesser extent, glucose and lactate. In DCM, heart failure, and ischemic heart disease, an increased relative ratio of mitochondrial fatty acid oxidation to carbohydrate oxidation in cardiomyocytes leads to decreased cardiac efficiency and disease progression, which might be associated with the overactivation of forkhead box protein O1 (FOXO1) [ 103]. Increasing cardiac glucose metabolism or inhibiting cardiac fatty acid oxidation has been shown to improve cardiac function and metabolic status.
Currently, many strategies that improve mitochondrial fatty acid metabolism in cardiomyocytes are available for the treatment of DCM, Beaudoin et al. have found that feeding resveratrol to obese type 2 diabetic rats (ZDF rats) decreases the accumulation of intracellular triglycerides, diglycerides, and ceramides, and decreases fibrosis, sensitivity to palmitoyl-CoA, and mitochondrial ROS emission rates, thus providing an effective strategy for preventing DCM [ 104]. Turkieh et al. have generated APOO-TG mice by expressing the apolipoprotein O (APOO) gene and observed a decline in cardiac function, and enhanced mitochondrial uncoupling and respiration, fatty acid metabolism, and ROS production. These effects were associated with increased AMPK phosphorylation and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PPARGC1α) expression, but were mitigated by targeting and knocking down APOO. Therefore, targeting APOO-dependent metabolic remodeling might serve as a strategy to adjust cardiac metabolism and protect the myocardium against contractile impairment [ 105]. Currently, traditional Chinese medicine plays important roles in the diagnosis and treatment of diseases, and its specific mechanisms are increasingly being explored. Shengmai San, a traditional Chinese medicine formulation made from ginseng, Ophiopogon japonicus, and Schisandra chinensis, are currently widely used for the treatment of ischemic diseases. Tian et al. have found that feeding Shengmai San at a dose of 3 g/kg body weight aids in recovery from diabetes-induced myocardial hypertrophy and diastolic dysfunction. Transmission electron microscopy has shown alleviation of cardiac lipid droplet accumulation and mitochondrial morphological damage, and further mechanistic studies have suggested that this treatment may act through the Sirt1/AMPK/PGC-1α signaling pathway [ 106]. Zhang et al. have found that cholesterol-25-hydroxylase (CH25H) ameliorates mitochondrial function and structural damage, as well as lipid accumulation in mice with type 2 diabetes, and is associated with the activation of PGC-1α [ 107]. Recently, in vitro experiments have demonstrated that a new oral glucagon-like peptide-1 receptor agonist, oral hypoglycemic peptide 2 (OHP2), prevents palmitic acid-induced oxidative stress and mitochondrial dysfunction by inhibiting intercellular lipid accumulation in cardiomyocytes, and therefore might provide a promising oral GLP-1 receptor agonist for preventing DCM [ 108]. As described above, MST1 knockout or inhibition improves mitochondrial dynamics and autophagy in DCM. Recent studies have indicated that Mst1 knockdown decreases lipotoxic cell apoptosis and inflammatory responses. Further research has shown that Mst1 expression under lipotoxic stimulation is regulated by FOXO3. Mst1 downregulation protects db/db mice against lipotoxic cardiac damage by inhibiting mitogen-activated protein kinase kinase kinase 1/c-Jun N-terminal kinase (MEKK1/JNK) signaling; therefore, Mst1 knockdown might be a potential strategy for treating DCM in type 2 diabetes patients [ 109]. Adipsin, an enzyme involved in the regulation of fat metabolism, is produced primarily by the body’s fat cells, where it helps break down triglycerides stores. Recent studies by Jiang et al. have shown that adipose tissue-specific overexpression of Adipsin significantly improves cardiac function in DCM rats, decreases the accumulation of lipids and their metabolites in the myocardium, alleviates cardiac remodeling, and enhances mitochondrial structural integrity and function. This mechanism is associated with interaction with interleukin-1 receptor-associated kinase-like 2 (Irak2) and inhibition of Irak2 mitochondrial translocation, thus promoting fatty acid β-oxidation in DCM [ 110].
Natural substances such as resveratrol, the traditional Chinese medicine Shengmai San, and glucagon-like peptide-1 receptor agonists (GLP-1RA) have been demonstrated to significantly ameliorate lipid metabolism disorders in cardiac mitochondria and to have substantial clinical application value. Additionally, Mst1 and Adipsin are research targets that reveal significant mechanisms.
Advanced Glycation End Products
AGEs are stable protein modification products produced under hyperglycemic conditions. In patients with diabetes, owing to prolonged hyperglycemia, proteins undergo non-enzymatic reactions with glucose and subsequently form AGEs. AGEs adversely affect the heart through various pathways, such as altering the extracellular matrix, activating inflammatory responses, increasing oxidative stress, affecting endothelial function, and promoting apoptosis. Myocardial mitochondria are also severely affected by AGEs, which lead to increased production of ROS within mitochondria, disruption of mitochondrial dynamics, effects on gene repair, and functional damage and dysregulation [ 111]. Zhang et al. have found that inhibiting the protein kinase C βII isoform (PKCβII) rescues methylglyoxal-AGE-induced oxidative stress, O(2−) production, cell death, apoptosis, and mitochondrial damage in rat cardiomyocytes. The mechanism may be associated with inhibition of NADPH oxidase and the alleviation of mitochondrial uncoupling [ 112]. Methylglyoxal, a byproduct of glycolysis and a precursor to the formation of AGEs, is significantly elevated in the diabetic myocardium. Recent studies have indicated that the mitochondrial-targeted methylglyoxal scavenger MitoGamide, administered via oral gavage, ameliorates diastolic dysfunction in the heart, although the specific mechanisms remain to be further explored [ 113]. Currently, AGE formation inhibitors being tested in clinical trials include aminoguanidine and alagebrium (ALT-711), which have not yet been widely applied.
Beneficial Effects of Exercise
Current treatment of diabetes relies on medication and dietary control; however, some patients still find achieving ideal glycemic control targets difficult. In recent years, exercise therapies such as swimming, running, aerobic exercise, yoga, Pilates, tai chi, and dancing have shown positive effects. Additionally, exercise effectively improves the structure and function of cardiac mitochondria by increasing mitochondrial quantity, enhancing mitochondrial efficiency, and improving mitochondrial dynamics, thereby enhancing cardiac function. In recent years, research on the mitochondrial mechanisms associated with exercise has become increasingly comprehensive.
Exercise has beneficial effects on cardiomyocyte apoptosis. Inhibiting the P2X7 purinergic receptor (P2X7R) has been shown to alleviate cardiac fibrosis and apoptosis in type 1 diabetes. Aerobic exercise reverses cardiac remodeling in diabetic mice, partly by inhibiting the expression of P2X7R in cardiomyocytes [ 114]. Wang et al. have found that exercise training significantly reverses the decrease in ejection fraction in db/db mice. Further mechanistic studies have revealed that exercise-induced activation of PGC-1α and Akt signaling significantly decreases myocardial cell apoptosis and fibrosis, the Bax/Bcl-2 ratio, and type I and III collagen levels; increases DNA (mtDNA) replication and transcription, and mtDNA content; and decreases mitochondrial ultrastructural damage [ 115].
Appropriate exercise effectively decreases ROS production. Veeranki et al. have further studied the effects of moderate-intensity exercise on DCM and observed recovery of connexin 43 (Cx43) levels in the interstitium and microvascular after exercise; improvements in gap junctional intercellular communication; and prevention of the decrease in the mitochondrial dynamics regulatory factor (Mfn2/Drp-1) ratio in the heart in db/db mice after exercise [ 85]. Additional studies have shown that resistance exercise training in rats increases the number of isolated mitochondria; enhances the expression of mitochondrial biogenesis proteins such as proliferator-activated receptor-γ coactivator-1α and TFAM; decreases proton leakage and ROS production; and results in elevated membrane potential. These findings are accompanied by increased superoxide dismutase 2, and decreased levels of UCP2 and UCP3, in agreement with previous research [ 116]. Wang et al. have further explored whether the mitochondrial improvement effects of exercise are associated with activation of the AMPK/PGC-1α axis, and have demonstrated that exercise protects against the shift in energy metabolism from fatty acid oxidation to glucose oxidation in diabetes. Depletion of PGC-1α weakens the mitochondrial function enhanced by exercise. Additionally, PGC-1α might be responsible for reversing the Warburg effect toward aerobic respiration, thereby enhancing mitochondrial metabolism and energy balance [ 117]. FGF21, a peptide hormone with multifaceted benefits in cardiac metabolic homeostasis, has been identified as an exercise-responsive factor. Recent research has indicated that exercise enhances the cardiac effects of FGF21 through AMPK-induced FOXO3 phosphorylation, induces expression of mitochondrial deacetylase SIRT3, and consequently reverses the hyperacetylation and functional damage to mitochondrial enzyme complexes induced by diabetes [ 118].
Exercise therapy, as an effective non-drug strategy for treating DCM, its mode of implementation, underlying mechanisms, and molecular targets mainly focuses on AMPK/PGC-1α/Sir3. However, the specific forms of exercise require further exploration and research.
Summary and Outlook
DCM is a complex metabolic heart disease with pathophysiological mechanisms involving multiple factors, including insulin resistance, oxidative stress, inflammatory responses, and mitochondrial dysfunction. In recent years, research has increasingly shown that mitochondrial dysfunction plays crucial roles in the occurrence and development of DCM. Dysfunction in mitochondria, the energy factories of cells, directly affects the bioenergetics of cardiomyocytes and promotes the development of DCM. Therefore, intervention strategies targeting mitochondrial dysfunction are becoming an important direction for DCM treatment. This review summarized current strategies used to improve or restore mitochondrial function through various mechanisms, such as regulating mitochondrial biogenesis, promoting mitophagy, decreasing mitochondrial uncoupling, using antioxidants, modulating the calcium balance, and optimizing lipid metabolism. These strategies significantly improve the energy metabolism and function of cardiomyocytes, and have shown potential therapeutic effects in preclinical and clinical studies ( Table 1). Nevertheless, many challenges remain to be overcome. First, the mechanisms underlying mitochondrial dysfunction may vary among patients, thus making the development of personalized treatment strategies particularly important. Second, many interventions targeting mitochondrial function face safety and efficacy issues in clinical applications; consequently, further research and validation are required. Additionally, integrating these emerging treatments with existing cardiovascular disease management strategies to achieve optimal therapeutic effects is another important direction for future research.
Summary of Targets or Drugs Alleviating Diabetic Cardiomyopathy
| Type | Drug/Target | Specific Mechanism | References |
|---|---|---|---|
| Calcium Regulation Disorder | Ferulic acid | SERCA/PLN | [ 10] |
| Rhynchophylline | Ryanodine receptor 2 phosphorylation | [ 11] | |
| MCU | -- | [ 12] | |
| Dihydromyricetin | -- | [ 16] | |
| reticulum–mitochondria Ca (2+)transfer | -- | [ 13] | |
| Mitofusin-2 | -- | [ 14] | |
| Bmal1 | Bcl2/IP3R | [ 15] | |
| Autophagy, Mitophagy, and Apoptosis | Eplerenone | -- | [ 19] |
| Synthetic liver X receptor agonist T0901317 | -- | [ 21] | |
| ALDH2 | AMPK | [ 25] | |
| Astragalus polysaccharides | -- | [ 22] | |
| FGF21 | -- | [ 23] | |
| Dihydromyricetin | -- | [ 24] | |
| Helix B surface peptide | AMPK | [ 25] | |
| MICU1 | mitochondrial Ca(2+) | [ 26] | |
| Exogenous H 2S | AMPK/mTOR/USP8 Sulfhydration | [ 27– 29] | |
| SIRT3 | Foxo3A-Parkin | [ 31] | |
| Polydatin | SIRT3 | [ 32] | |
| Melatonin | SIRT1-PGC1α | [ 33] | |
| JQ1 | BRD4/PINK1/Parkin | [ 34] | |
| Ginseng Dingzhi decoction | -- | [ 36] | |
| Resveratrol | -- | [ 35] | |
| Mitochondrial Modification | O-GlcNAcylation | -- | [ 41] |
| Resveratrol | SIRT3/PGC-1α | [ 42, 43] | |
| Adropin | GCN5L1 | [ 44] | |
| Lentinan | CAV1/SDHA | [ 45] | |
| Mitochondrial Oxidative Stress | mPHGPx | -- | [ 49] |
| Fasudil | -- | [ 50] | |
| GLP-1R | -- | [ 51] | |
| Mito-TEMPO | ERK1/2 | [ 52] | |
| ALDH2 | NLRP3 | [ 53] | |
| Calpain | ATP5A1 | [ 54] | |
| NAC | Endoplasmic reticulum stress | [ 56] | |
| DHA | -- | [ 57] | |
| Tom70 | -- | [ 58] | |
| NM-aFGF-loaded PEG-nanoliposomes | AKT/GSK-3β1/Nrf-2 | [ 59] | |
| Flavonoid | -- | [ 60] | |
| β-Lapachone | NQO1 | [ 61] | |
| 11-Keto-β-boswellic Acid | AMPK | [ 62] | |
| Macrod1 | PARP1-NAD-SIRT3 | [ 63] | |
| Dihydromyricetin | RIPK3-CaMKII | [ 64] | |
| FNDC5/Irisin | αV/β5-AKT | [ 65] | |
| LncDACH1 | SIRT3 | [ 66] | |
| MiR-340-5p | Mcl-1 | [ 67] | |
| MtDNA | Ogg1 | 8-OHdG | [ 73] |
| N-Acetyl cysteine, selenium, and ascorbic acid | PGC-1α, MFN2, Drp1 | [ 74] | |
| Mitochondrial Respiratory Chain Uncoupling | Protein lysine acetylation | -- | [ 76] |
| Methylene blue | Mitochondrial lysine acetylation | [ 78] | |
| Dihydromyricetin | Enhanced complex I/II/III/IV/V activity | [ 24] | |
| UCPs | -- | [ 79] | |
| Akap1 | NDUFS1 | [ 80] | |
| BGP-15 | TCA/ETC | [ 81] | |
| Mitochondrial Dynamics | GLUT1 | -- | [ 84] |
| Moderate intensity exercise | Cx43, Cx37 | [ 85] | |
| High intensity interval training | -- | [ 86] | |
| Resveratrol | SIRT1 | [ 87] | |
| Melatonin | SIRT1-PGC1α, SIRT6 | [ 33, 85] | |
| Rosmarinic acid | SIRT1/PGC-1α | [ 88] | |
| Ophiopogonin D | -- | [ 89] | |
| CaSR | Activation of the gp78-ubiquitin proteasome system | [ 90] | |
| Mst1 | Drp1 | [ 91] | |
| Mitochondrial fusion promoter | Opa1 | [ 92] | |
| Punicalagin | Opa1-PTP1B-Stat3 | [ 93] | |
| Nicotinamide | SIRT1-PGC1α-PPARα | [ 95] | |
| Empagliflozin | -- | [ 96] | |
| Salidroside | SIRT3 | [ 97] | |
| SFRP2 | -- | [ 98] | |
| Ketone ester D-β-Hydroxybutyrate-(R)-1,3 | -- | [ 99] | |
| MiR4 | Mitochondrial biogenesis | [ 100] | |
| BH4 | CaMKK2 | [ 101] | |
| Ameliorative Effects of Exercise | Exercise | PGC-1α-Akt | [ 115] |
| Moderate intensity exercise | Mfn2/Drp1 | [ 85] | |
| Resistance exercise | -- | [ 116] | |
| Exercise | AMPK/PGC-1α | [ 117] | |
| Exercise | FGF21-SIRT3 | [ 118] | |
| Aerobic exercise | P2X7R | [ 114] | |
| Mitochondrial Lipid Metabolic Disorder | Resveratrol | -- | [ 104] |
| Apolipoprotein O | AMPK/PPARGC1A | [ 105] | |
| Shengmai San | SIRT1/AMPK/PGC-1α | [ 106] | |
| CH25H | PGC-1α | [ 107] | |
| OHP2 | GLP-1RA | [ 108] | |
| Mst1 | MEKK1/JNK | [ 109] | |
| Adipsin | Irak2 | [ 110] | |
| Advanced Glycation end Products | PKCβII | NADPH oxidase inhibition and alleviation of associated mitochondrial uncoupling | [ 112] |
| MitoGamide | -- | [ 113] |
SERCA/PLN: sarcoplasmic reticulum Ca(2+)-ATPase/phospholamban; Bcl2/IP3R: B-cell lymphoma 2/Inositol 1,4,5-trisphosphate receptor; AMPK: adenosine 5′-monophosphate-activated protein kinase; mTOR/USP8: mammalian target of rapamycin/Ubiquitin Specific Peptidase 8; Foxo3A: Forkhead box O3A; BRD4: Bromodomain-containing protein 4; GCN5L1: General control of amino acid synthesis 5 like 1; CAV1/SDHA: Caveolin-1/Succinate dehydrogenase complex subunit A; JQ1: JQ-1 carboxylic acid; GLP-1R: glucagon-like peptide-1 receptor agonist; Mito-TEMPO: Mitochondria-targeted 2,2,6,6-tetramethylpiperidine-1-oxyl; ERK1/2: Extracellular Signal-Regulated Kinases 1 and 2; NLRP3: NOD-, LRR- and pyrin domain-containing protein 3; Bmal1: Brain and Muscle ARNT-Like 1; FGF21: Fibroblast Growth Factor 21; ATP5A1: ATP Synthase F1 Subunit Alpha 1; NAC: N-acetylcysteine; DHA: docosahexaenoic acid; Tom70: Translocase of Outer Mitochondrial Membrane 70; AKT: Protein Kinase B (also known as AKT kinase); GSK-3β1: Glycogen Synthase Kinase 3 β 1; Nrf-2: Nuclear Factor Erythroid 2-Related Factor 2; NQO1: NAD(P)H Quinone Dehydrogenase 1; PARP1: Poly(ADP-Ribose) Polymerase 1; NAD: nicotinamide adenine dinucleotide; RIPK3: Receptor Interacting Protein Kinase 3; CaMKII: Calcium/Calmodulin-Dependent Protein Kinase II; αV/β5: Integrin alpha V/β 5; AKT: Protein Kinase B; UCPs: Uncoupling Proteins; Akap1: A-Kinase Anchoring Protein 1; TCA: tricarboxylic acid (cycle), also known as the Krebs cycle or citric acid cycle; ETC: electron transport chain; Cx43: Connexin 43; Cx37: Connexin 37; Stat3: Signal Transducer and Activator of Transcription 3; P2X7R: P2X7 Receptor; IRAK2: Interleukin-1 Receptor-Associated Kinase 2.
In summary, interventions targeting mitochondrial dysfunction provide new hope and directions for ameliorating DCM. With deepening understanding of mitochondrial biology and pathological mechanisms, and the continued emergence of new technologies and therapies, we believe that precise regulation of mitochondrial function will bring more effective treatments to patients with DCM, and ultimately improve their quality of life and prognosis. Future research should continue to explore and validate the clinical application potential of these intervention strategies to further advance DCM treatment.
