
Introduction
RNA splicing is a widespread post-transcriptional process observed in eukaryotes, particularly in multi-exon genes. A single gene can produce multiple mature transcripts through alternative splicing (AS), thereby enhancing the human transcriptome’s overall complexity. This process is crucial for fine-tuning cellular identity and function. The regulation of AS is crucial for cellular specificity and is governed by cis-regulatory enhancers and silencers within pre-mRNAs, which interact with trans-acting splicing factors. Dysregulated AS plays a major role in various human diseases.
For decades, cardiovascular disease (CVD) has been a leading cause of morbidity and mortality worldwide, thus posing a substantial burden on healthcare systems. Despite intensified research endeavors in CVD, the diagnosis, management, and treatment of these conditions continue to face major challenges. Dysregulated AS has emerged as a key mechanism underlying many aspects of CVD. This review aims to offer a comprehensive examination of AS and its regulatory mechanisms within the cardiovascular system (Figure 1).

Description of RNA Splicing
RNA splicing requires the spliceosome, whose formation involves a complex interplay of trans-acting factors, including five small nuclear ribonucleoprotein particles (snRNPs): U1, U2, U4/U6, and U5 snRNPs. Each of these snRNPs consists of snRNAs and associated proteins (Figure 2). The spliceosome assembly involves approximately 150 proteins.
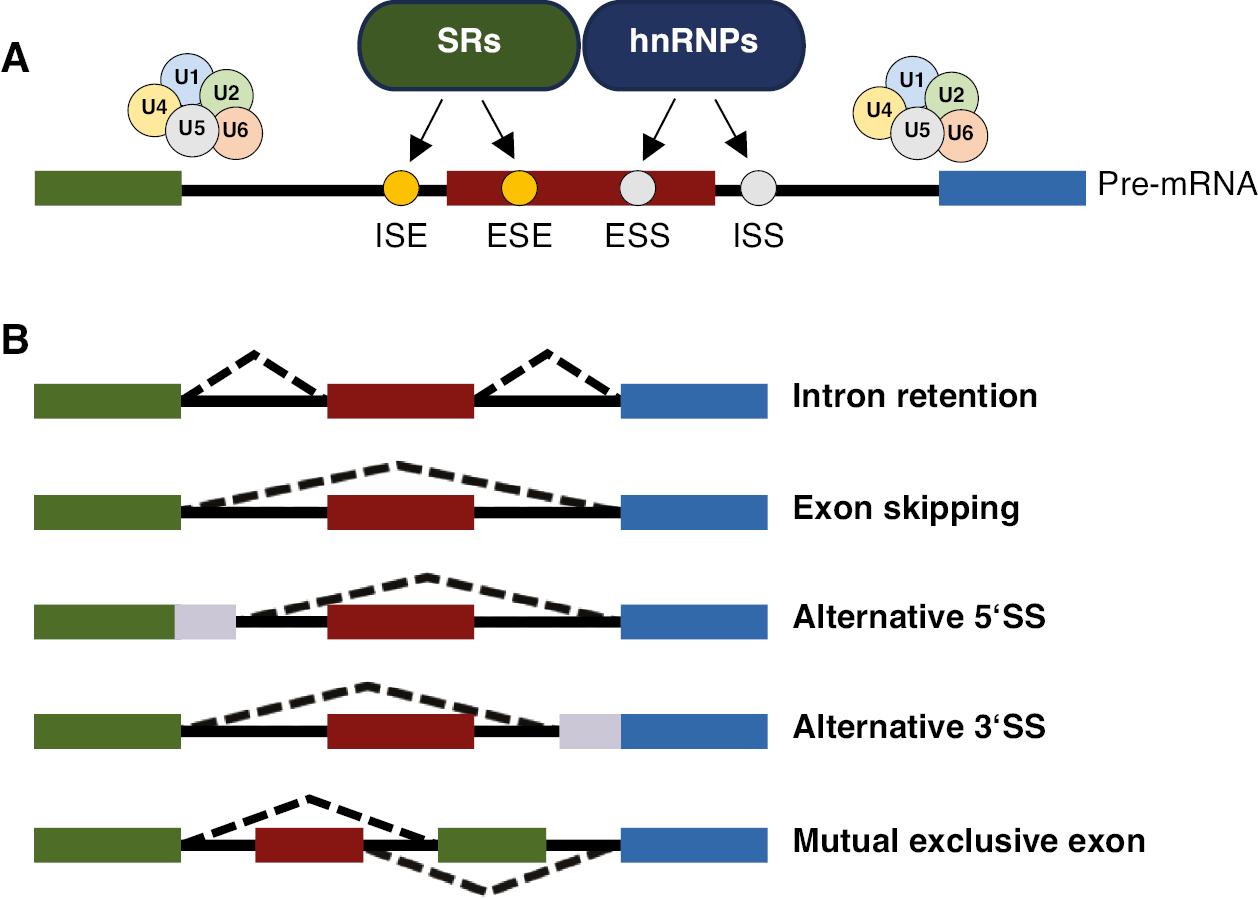
Alternative Splicing and its Regulation.
(A) Regulatory sequences and factors in alternative splicing. Splicing is regulated by cis-regulatory sequences (including exonic splicing enhancers, ESSs, ISEs, and intronic splicing silencers), and two main families of splicing factors (including SRs and hnRNPs). Splicing factors targeting components of spliceosome that associate with both the 5′ SS and the 3′ SS and can activate or inhibit the recognition and use of that site, thus mediating exon definition. Splicing is performed by a large ribonucleoprotein complex (spliceosome) composed of more than 100 core proteins and five snRNAs (U1, U2, U4, U5, and U6). (B) Simplified model of alternative splicing. Alternative splicing adds a layer of complexity to gene expression regulation, allowing a single gene to produce multiple RNA isoforms, including mutually exclusive exons (MXE), intron retention (IR), exon skipping (ES), and alternative 5′SS and 3′SS (A5SS/A3SS).
The procedure of RNA splicing involves sequential binding of snRNPs to form the spliceosome, which performs two trans-esterification reactions that complete RNA splicing. Current models suggest that U1 snRNP first binds the 5′ splice site (5′ SS) and interacts with SF1 and the U2 snRNP auxiliary factors (U2AF1 and U2AF2) at the 3′ splice site (3′ SS) and its adjacent polypyrimidine tract through bridging interactions. SR proteins and SR-related proteins – each containing one or more intrinsically disordered regions rich in alternating arginine and serine residues, known as RS domains – mediate additional interactions that facilitate exon and intron definition. These early interactions aid in the stable recruitment of U2 snRNP to the pre-mRNA branch site, which is followed by the addition of U4/U6 and U5 snRNPs, and the formation of a tri-snRNP particle. The actions of various RNA helicases subsequently promote rearrangements of snRNP interactions that establish a catalytically competent spliceosome capable of performing the two trans-esterification reactions leading to lariat formation, intron removal, and exon ligation.
Various RNA-binding proteins (RBPs) regulate AS, including the SR proteins and the heterogeneous ribonucleoprotein (hnRNP) family, as well as RBPs containing domains such as RNA recognition motif (RRM), K homology (KH), zinc-finger, and others. Cis-acting elements, such as exonic splicing enhancers, exonic splicing silencers (ESS), intronic splicing enhancers (ISE), and intronic splicing silencers located in introns or exons, positively or negatively mediate splicing. The mechanisms of AS (illustrated in Figure 2) encompass four main modes: exon skipping (ES), mutually exclusive exon usage (MXE), alternative 5′SS/3′SS selection (A5SS/A3SS), and intron retention (IR).
Screening and Validation Methods for Alternative Splicing Events
AS plays crucial roles in expanding the diversity of gene expression and is involved in numerous physiological processes and diseases. The detection and validation of AS events are essential for understanding their regulatory mechanisms and potential effects on human health. Frequently used methods can be broadly categorized into high-throughput screening techniques and specific validation strategies. Key technologies and approaches are described below.
RNA-Seq and Iso-Seq (Full-Length Isoform Sequencing)
RNA sequencing (RNA-Seq) is the most widely used high-throughput technique for detecting AS events. By capturing and sequencing the entire transcriptome, RNA-Seq can identify different splice variants across various conditions, tissues, or developmental stages [1, 2]. Iso-Seq, a full-length transcript sequencing technology, offers a more accurate representation of splice variants by sequencing complete transcripts without fragmentation. Iso-Seq provides an invaluable tool for capturing low-abundance isoforms and obtaining high-resolution maps of AS events.
CLIP-Seq
Crosslinking immunoprecipitation sequencing (CLIP-seq) is an RBP-targeting technique used to identify binding sites of splicing factors on pre-mRNAs. Enhanced CLIP-seq (eCLIP-seq), an advanced version of CLIP, provides enhanced specificity and reproducibility, and enables comprehensive profiling of splicing factor interactions [3]. This method is instrumental in discovering the regulatory networks involved in AS by linking splicing factors to specific alternative exons and introns.
Splicing-Sensitive Microarrays
Splicing-sensitive microarrays are designed to detect known splicing events by probing specific exon-exon junctions and splice sites. Although currently less commonly used than RNA-Seq, microarrays provide a cost-effective, efficient, and high-throughput alternative for screening a predefined set of AS events in large cohorts. They are useful in studies focused on well-characterized AS patterns.
Validation Methods for Alternative Splicing Events
CRISPR/Cas9 genome editing has been used to precisely manipulate specific splicing events in cells by knocking in mutations that alter splice sites or regulatory sequences. This method is highly effective for studying the molecular mechanisms of AS, because it allows researchers to observe the functional consequences of specific splicing variants in living cells. Moreover, this method is particularly valuable in determining the roles of AS events in disease contexts.
Advances in big data and cohort studies have enabled the discovery of disease-associated AS events. By analyzing RNA-Seq data from large populations or patient groups, researchers can identify splicing variations associated with specific diseases. This approach not only facilitates the detection of biomarkers but also aids in the development of therapeutic targets.
Artificial Intelligence-based Models
Artificial Intelligence (AI)-based models are increasingly used for predicting and analyzing AS. These models incorporate genomic, transcriptomic, and epigenetic data to predict AS patterns according to sequence features and splicing factor binding motifs. Machine learning approaches can efficiently sift through large datasets to uncover novel splicing events and prioritize disease-relevant AS changes for further study.
Luciferase Reporter Assays
Luciferase reporter assays are widely used for the functional validation of AS events. By cloning specific exons or introns of interest into a reporter construct, researchers can quantitatively measure the splicing efficiency or exon inclusion through changes in luciferase activity. This method is particularly useful for studying the regulatory elements that control splicing decisions.
Northern Blot Analysis
Northern blot analysis, a classic method for validating specific AS events, enables detection of different mRNA isoforms by separating RNA on a gel, transferring it to a membrane, and probing the membrane with labeled RNA or DNA probes specific to alternative exons. Although this method is infrequently used because of its lower sensitivity than newer techniques, Northern blotting provides a visual representation of the splicing pattern and can serve as a reliable validation method for known splicing variants.
These techniques, when used in combination, provide a comprehensive toolkit for identifying, validating, and characterizing AS events, and consequently advancing understanding of gene regulation and its implications for human health.
Alternative Splicing Regulates Sarcomere Contraction
AS of sarcomeric genes is among the earliest processes associated with both acquired and inherited heart diseases. Sarcomeres constitute the fundamental contractile units within striated muscle fibers, comprising contractile, structural, and regulatory proteins (Table 1). Actin (thin filaments) and myosin (thick filaments) are central to muscle contraction. The mechanism of filament sliding is propelled by cross-bridges, wherein myosin heads extend from the myosin backbone to cyclically interact with actin filaments (Figure 3).
Summary of Sarcomere Genes Regulated by AS.
| Gene | AS | Functional effect | Disease | Reference |
|---|---|---|---|---|
| TTN | IR | Shift from less compliant N2B toward highly compliant N2BA isoforms | DCM | [4] |
| TPM | IR | Less structural stability and weaker actin-binding affinity | DCM, HF | [5] |
| TNNT2 | ES | Ca2+ sensitivity | HF | [6] |
| MYH7 | ES | Premature stop codons cause nonsense-mediated decay | ICM | [7] |
| FLNC | IR | Altered structure or function of Z-disks | DCM | [7] |
| TNNI3 | IR | Premature stop codons | HF | [7] |
| Myomesin | IR | Adaptive remodeling of the sarcomere cytoskeleton in the dilated heart | DCM, HCM | [8] |
| LDB3 | ES | Dysregulation of Ca2+ homeostasis | DCM | [9] |
| ENH | IR | Premature stop codon | HCM | [10] |
| Nebulin | ES | Impaired Nebulin-tropomyosin interaction along the thin filament | Myopathy | [11] |
| KCNQ1 | A5SS | Differential IKs amplitude | Arrhythmia | [12] |
| CaV1.2 | A5SS | Inactivation | Hypertension | [13] |
| SERCA2 | A3SS | Different enzymatic properties | HF | [14] |
| RyR2 | IR | Intracellular Ca2+ signaling | Cardiomyocyte (CM) apoptosis | [15] |
| CaMKIIδ | IR | Ca2+/calmodulin transportation | Hypercontraction | [16] |
| SCN5A | ES | Cardiomyocyte structural changes | DM | [17] |
| Nav1.5 | MXE | Ion conduction and voltage-dependent gating | DM | [18] |
| IGF-1 | IR | Nuclear localization | MI | [19] |
| VEGFA | A3SS | Singal transduction | DCM | [20] |
| TBX5 | IR | Binding to SC35 | HOS | [21] |
| XBP1 | ES | ER stress | IRI | [22] |
| Myocardin | IR | Endothelial mesenchymal transition (EndMT) | Cardiogenesis | [23] |
| MYH7b | IR | Prevention of protein expression | Cardiomyopathies | [24] |
| CARMEN | A5SS A3SS | Specification in cardiomyocytes and the smooth muscle cell (SMC) lineage | Cardiogenesis | [25] |
| Snhg15 | NA | Interaction with alternative splicing regulator Zranb2 | HF | [26] |
| lncRNA | NA | Regulation of AS | MI | [27] |
Note: ES, exon skipping; MXE, mutually exclusive exon usage; A5SS/A3SS, alternative 5′SS/3′SS selection; IR, intron retention; ICM, ischemic cardiomyopathy; DM, myotonic dystrophy; MI, myocardial infarction; HOS, Holt-Oram syndrome; DCM, dilated cardiomyopathy; HF, heart failure; IRI, ischemia-reperfusion injury.
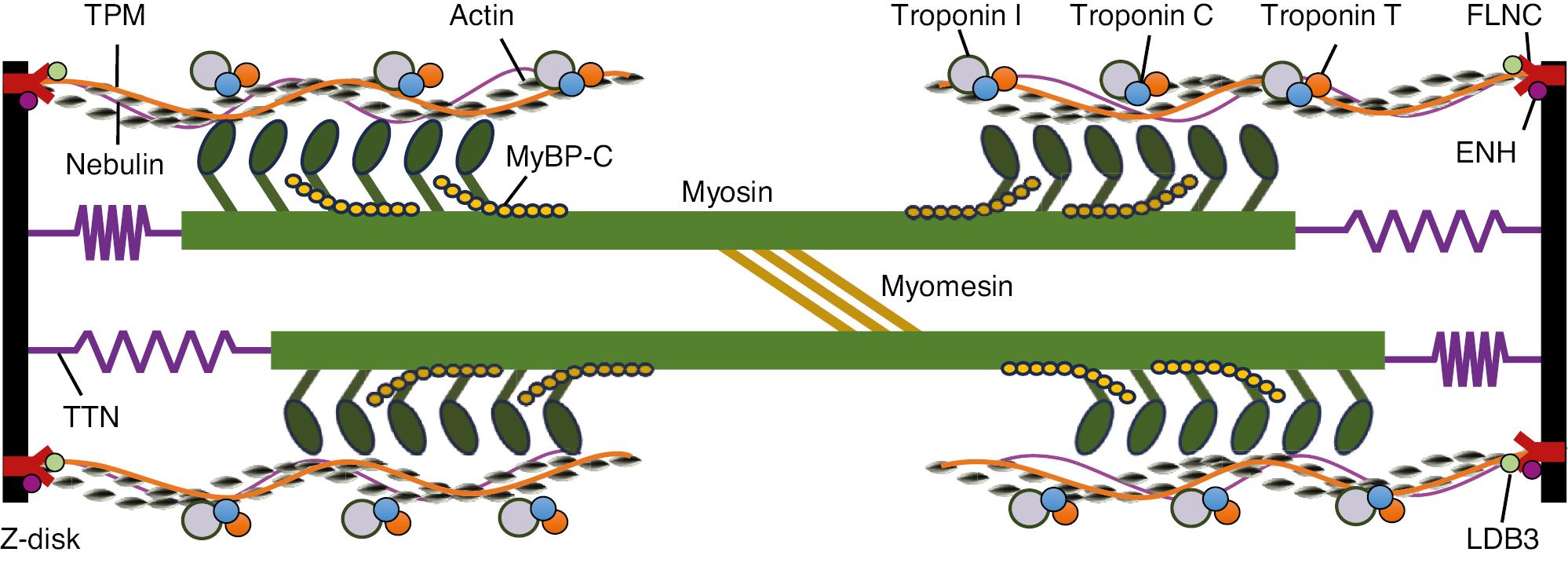
Schematic Illustration of a Portion of the Sarcomere, the Contractile Unit of Muscle, Composed of Thick and Thin Filaments.
The thin filaments contain actin, tropomyosin (TPM), Nebulin, troponins C (TNNC), I (TNNI), and T (TNNT). The thick filaments are composed of myosin (MYH7) and MyBP-C, with the globular heads forming cross-bridges with thin filaments. The giant protein TTN extends along the length of an entire half-sarcomere. The M-line contains various proteins, such as myomesin, whereas LDB3, ENH, and FLNC are located in the Z-discs. Of note, the illustration does not correspond to actual size.
TNNT2 encodes the cardiac muscle isoform of troponin T (cTnT), situated on the thin filament of striated muscles, which plays crucial roles in regulating muscle contraction in response to fluctuations in intracellular calcium ion levels. A mutation in the splice donor site of intron 15 of TNNT2 leads to protein truncation and is associated with familial hypertrophic cardiomyopathy [28, 29]. MBNL1 plays crucial roles in regulating the splicing of exon 5 in TNNT2 pre-mRNA by directly competing with the essential splicing factor U2AF65 for binding at the 3′ end of intron 4. This competition disrupts the recruitment of U2 snRNP, such that U2AF65 is unable to bind the pre-mRNA, and the subsequent exon is ultimately skipped. Furthermore, MBNL1 and U2AF65 compete by binding distinct RNA structures that are mutually exclusive. Specifically, at the 3′ end of intron 4, splicing factors can use either a stem-loop or a single-stranded structure. MBNL1 preferentially binds the intron in a stem-loop conformation, whereas U2AF65 binds the same region in a single-stranded conformation. Exon 5 of cTnT encodes a highly acidic 9- or 10-amino acid segment, which carries a negative charge at physiological pH. It is expressed in the embryonic heart but is downregulated and eventually ceases to be expressed during postnatal development. The embryonic form of cardiac troponin T (cTnT), with a more negatively charged N-terminal region, confers greater calcium sensitivity to actomyosin ATPase activity and myofilament force production than the adult form of cTnT. Additionally, the embryonic form exhibits a higher tolerance to acidosis. The switch between isoforms, as a consequence of AS of exon 5 of TNNT2, is also subject to regulation by CUGBP2 and SRSF1 [30, 31].
Myosin-binding protein C (MyBP-C), a key regulatory protein within sarcomeres, is arranged transversely in the A-band, where it interacts with myosin heavy chains and titin. Whereas MyBP-C has several isoforms expressed in different muscle types, the cardiac isoform (cMyBP-C) plays crucial roles in modulating cardiac muscle contraction.
Unlike the skeletal isoforms, cMyBP-C contains unique phosphorylation sites that are critical for regulating the dynamics of sarcomere function, particularly during the cardiac cycle. Phosphorylation of cMyBP-C affects its interaction with actin and myosin, thereby fine-tuning the force and speed of cardiac contraction.
Furthermore, hundreds of MYBPC3 variants (MYBPC3 is the gene that codes for the cMyBP-C protein), associated predominantly with hypertrophic cardiomyopathy (HCM), have been identified in cMyBP-C. These include conserved RNA splice site mutations that result in truncated polypeptides. The production of these truncated proteins increases their degradation, and consequently decreases the overall levels of cMyBP-C in the sarcomere and results in haploinsufficiency. This decrease in cMyBP-C disrupts normal cardiac contractility and contributes to the pathophysiology of HCM [32, 33].
Tropomyosin (TPM), a critical component of thin filaments in muscle cells, plays crucial roles in regulating the interaction between actin and myosin, which is essential for muscle contraction. AS of TPM generates multiple gene variants, which are often specific to different cell types. In the heart, AS of exon 2b in TPM1 generates a variant associated with human DCM and HF [5]. The TPM1κ isoform, one of ten distinct products originating from the TPM1 gene, enables modulation of sarcomeric performance in response to conditions such as exercise, stress, or cardiac disease. AS of exon 2b in the TPM1 gene gives rise to the TPM1κ isoform, which constitutes approximately 50% of striated muscle TPM1 mRNA. The TPM1κ protein is expressed and integrated into organized myofibrils within cardiac tissue, and its levels are elevated in DCM and HF. Heightened incorporation of TPM1κ protein into myofilaments has been implicated in the development of DCM. Further biophysical investigations have revealed that TPM1κ exhibits lower structural stability and weaker actin-binding affinity than the TPM1α isoform.
The TTN (Titin) gene encodes the Titin protein, which acts as a molecular spring spanning half of the sarcomere and linking the Z-disk to the M-line. AS of TTN is closely associated with DCM and familial hypertrophic cardiomyopathy. Approximately one-third of Titin mutations identified in individuals with familial DCM affect splicing processes. These mutations lead to exon skipping and intron retention, and result in truncated titin isoforms and the development of DCM. Titin splicing is regulated by the RNA-binding protein RBM20, which is expressed predominantly in cardiac tissue. RBM20 plays a key role in controlling the AS of genes associated with diverse cellular processes (Figure 4). In rats, the absence of RBM20 leads to abnormal Titin splicing, cardiac fibrosis, arrhythmias, diastolic dysfunction, and DCM, which together mirror the clinical phenotype observed in humans with RBM20 mutations [34]. Moreover, TTN produces a class of circular RNAs (cTTN1) that rely on RBM20 for their formation [35]. LDB3, located in the Z-disc, plays crucial roles in maintaining the sarcomeric structure. Dysfunction in RBM20 results in the exclusion of exon 4 from mature LDB3 mRNA [34].
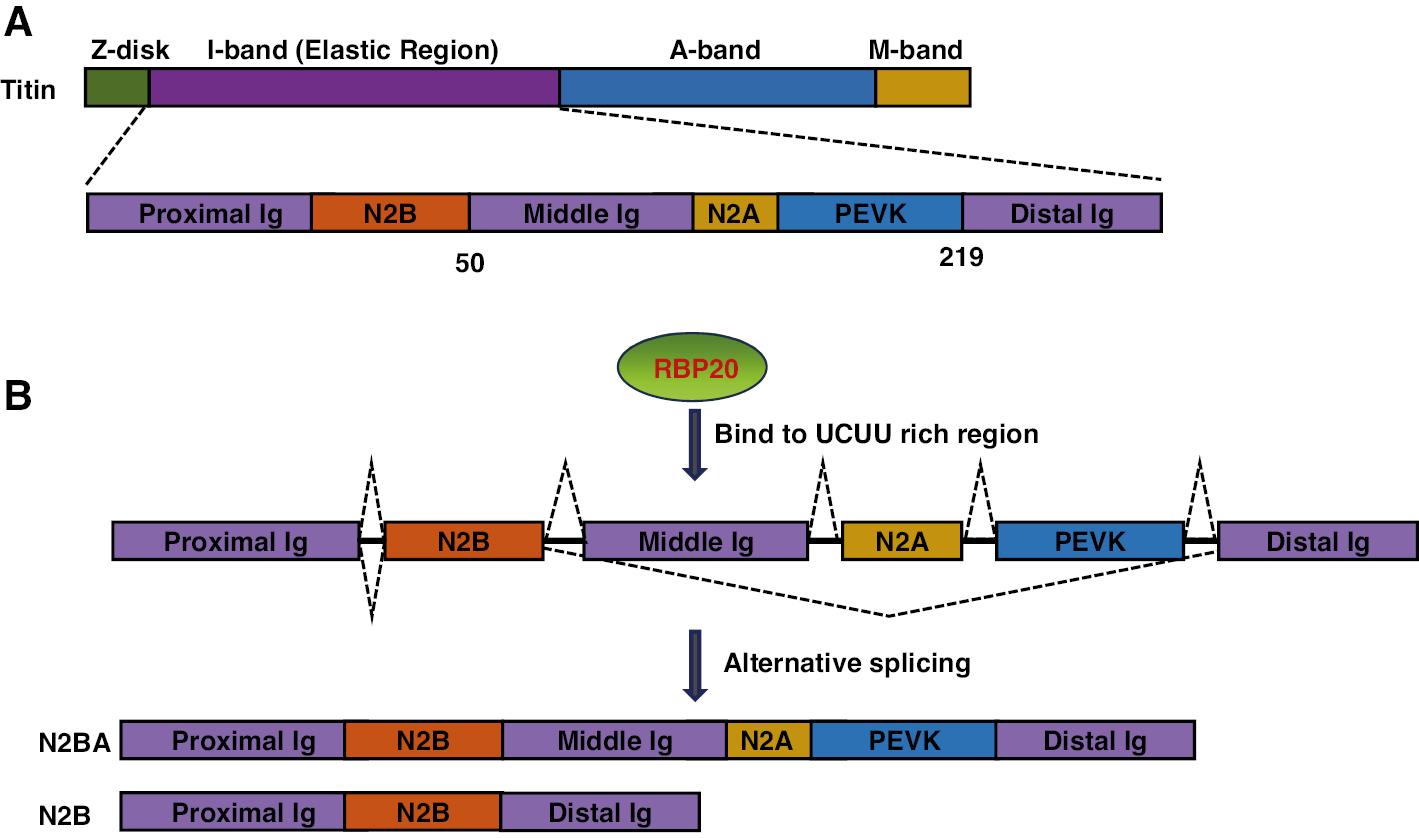
Structural Representation of Titin Protein Isoforms.
(A) Schematic diagram of titin’s domain organization, with domain names and positions indicated. The corresponding exons are shown below each domain. Key domains include the distal Ig repeat domain, middle Ig repeat domain, proximal Ig repeat domain, and the unique N2A and N2B elements. (B) Schematic illustration of the titin isoforms, highlighting the alternatively spliced regions, with dotted lines indicating areas of high variability.
EH-myomesin, also known as Myomesin-2 or MYOM2, plays crucial roles in maintaining the structural integrity of sarcomeres and organizing thick filaments, particularly within the M-band region. An alternatively spliced version of EH-myomesin has been identified as a marker for HF. The predominant splice isoform of EH-myomesin in the embryonic heart is markedly upregulated in failing hearts, in correlation with decreased cardiac function. Quantitative RT-PCR analysis has demonstrated a 41-fold elevation in the EH-myomesin isoform in patients with DCM compared with controls. Immunofluorescence analyses have revealed a cell-specific enhancement of EH-myomesin that results in increased heterogeneity of the myocytes’ cytoskeleton across the myocardial wall. The upregulation of EH-myomesin reflects adaptive remodeling of the sarcomere cytoskeleton in dilated hearts and might serve as a marker for DCM in both mouse and human myocardium [8].
ENH interacts with sarcomere components and significantly contributes to muscle fiber organization and stability; it is expressed in the heart, as well as various other tissues. The PDZ domain of ENH1 binds α-actin, which subsequently localizes at the Z-disc in neonatal rat cardiomyocytes. Through AS, ENH generates at least four isoforms: ENH1, ENH2, ENH3, and ENH4. Among these isoforms, ENH1 uniquely contains all three LIM motifs at the C-terminal domain. These splice variants exhibit differential expression patterns in embryonic, neonatal, and adult heart tissues. ENH1 predominates in the embryonic heart, whereas the adult heart expresses primarily the truncated isoforms ENH2, ENH3, and ENH4. In mice, hypertrophy induced by transaortic constriction causes the expression pattern of ENH isoforms in the heart to revert to a neonatal/embryonic profile [36] Moreover, overexpression of ENH1 in neonatal cardiomyocytes promotes hypertrophy, whereas silencing of ENH1 prevents hypertrophy. In contrast, overexpression of ENH4 inhibits the development of hypertrophy. These findings suggest that AS of ENH transcripts generates antagonistic isoforms during heart development: the embryonic isoform ENH1 may induce hypertrophy, whereas the adult isoform ENH4 may protect against cardiac hypertrophy [37].
The advent of next-generation sequencing has significantly enhanced understanding of transcriptome-wide alterations and revealed several overarching splicing patterns. A comprehensive assessment of mRNA splicing in left ventricular myocardial RNA from control individuals and patients with ICM through Affymetrix Exon array analysis has indicated a marked and widespread decrease in mRNA splicing efficiency in HF. This decrease affects certain introns more profoundly than others.
Furthermore, AS of four crucial sarcomere genes – cardiac troponin T (TNNT2), cardiac troponin I (TNNI3), myosin heavy chain 7 (MYH7), and filamin C, gamma (FLNC) – is significantly modified in ICM, DCM, and aortic stenosis [7]. In a study by Giudice et al., mRNA deep sequencing (RNA-Seq) has been used to analyze AS and gene expression transitions in freshly isolated cardiac fibroblasts and cardiomyocytes during development. Most gene expression and AS changes were found to occur within the initial 4 weeks after birth [38]. Furthermore, Wang et al. have discovered distinct AS patterns in fetal and adult hearts through RNA-Seq data analysis. Essential genes such as CACNB2, TPM1a, MBNL1, cTNT, and Dab1 exhibit conserved splicing transitions across species during heart development. Additionally, MYH6 and MYH7 show differential expression patterns, with MYH7 predominating in late fetal life and transitioning to MYH6 after birth, in agreement with adult expression patterns [39].
Nebulin plays crucial roles in assembly of the contractile machinery within muscle cells. Mutations in the NEB gene, which encodes Nebulin, are a common cause of nemaline myopathy. AS of nebulin mRNA yields two distinct mRNA variants containing either exon 143 or exon 144. The isoform of nebulin containing exon 144 is the primary from early in myogenesis, whereas the expression of the isoform containing exon 143 is regulated and occurs during later stages of muscle development [11].
Alternative Splicing Regulates Ion Channel Function
The regulation of calcium entry through the cell membrane involves voltage-dependent L-type calcium channels, which are composed of α1, α2/δ, β, and γ subunits. Within the heart, the primary α1 subunit (Cav1.2) undergoes AS across various exons during both development stages and in HF. This AS process generates a subset of Cav1.2 channel splice variants that include exon 9 and play crucial roles in maintaining vasotone in blood vessels [40].
Calcium handling in the sarcoplasmic reticulum involves AS of both calcium ATPase 2 (SERCA2) and the ryanodine receptor (RyR) mRNAs. RyR2, a crucial regulator of cardiac function, exists in two alternatively spliced variants characterized by 30- and 24-bp sequence insertions. The 24-bp variant (encoding the peptide sequence VTGSQRSK) and the 30-bp variant (encoding the peptide sequence FAIDSLCGFG) of RyR2 represent two alternatively spliced forms. These variants have been suggested to play distinct roles in cardiac function and signaling pathways. Specifically, the expression of these variants in cardiomyocyte cell lines significantly modulates cellular architecture and calcium (Ca2⁺) signaling.
Notably, the 24-bp variant has been associated with a protective effect against stimulation-induced apoptosis in cardiomyocytes. Therefore, this variant might enhance cellular resilience under stress conditions, whereas the functional implications of the 30-bp variant remain less defined. Further investigations are necessary to fully elucidate the distinct roles of these variants in cardiac development and their effects on cell signaling pathways [15]. The splicing of RyR is regulated by SRSF2, a trans-regulatory factor. The induction of SRSF2 has been proposed to serve as a compensatory mechanism in patients with cardiac hypertrophy due to aortic stenosis [41].
SERCA2 pumps calcium back into the sarcoplasmic reticulum and consequently facilitates muscle relaxation. The expression of the SERCA2a isoform in cardiac and slow skeletal muscle is governed by AS. In homozygous SERCA2a−/− mice, the absence of SERCA2a mRNA and protein results in compensatory upregulation of the SERCA2b isoform. However, the levels of SERCA2b mRNA and protein in the hearts of SERCA2a−/− mice reach only 50% of the total SERCA2 expression observed in wild-type mice. Consequently, these mice exhibit diminished calcium uptake, impaired cardiac contractility and relaxation, developmental abnormalities in the heart, and mild concentric hypertrophy [36].
CaMKIIδ phosphorylates various calcium-handling proteins, including the SERCA regulator phospholamban. The postnatal isoform transition of CaMKIIδ is mediated by the trans-regulatory factors SRSF1 and RBM20. Loss of either factor leads to the exclusion of exon 14, which encodes a nuclear localization signal, and consequently results in protein mislocalization. SRSF1 knockout mice exhibit severe excitation-contraction coupling defects, cardiac fibrosis, and premature mortality at 6–8 weeks of age. These defects are phenotypically mimicked by the transgenic expression of alternatively spliced CaMKIIδ in cardiomyocytes; therefore, the AS of this kinase might contribute to the observed phenotype in SRSF1 knockout mice [16].
The SCN5A gene encodes the alpha subunit of the voltage-gated sodium channel Nav1.5, which is expressed primarily in cardiomyocytes. Nav1.5 plays crucial roles in initiating and propagation action potentials in the heart, and contributes to the generation of the cardiac electrical signal essential for normal heart function. Mutations in the SCN5A gene are associated with various cardiac arrhythmias and conduction disorders, including long QT syndrome (LQTS), Brugada syndrome, and cardiac conduction disease. Explanted ventricles show upregulation of two SCN5A variants and downregulation of the full-length SCN5A mRNA transcript. These variants display diminished membrane protein expression and lack functional current [42]. Alterations in SCN5A splicing in these patients are associated with increased expression of splicing regulators RBM25 and LUC7L3 (Figure 5). These regulators favor the production of a truncated isoform and decreased levels of the full-length protein, thus resulting in a significant decrease in Na+ current [43]. Additionally, a switch in SCN5A splicing from the adult exon 6B to the fetal exon 6A in the cardiac sodium channel has been observed. Furthermore, MBNL1 has been identified as a regulator of AS of SCN5A mRNA. The splicing variant of SCN5A observed in patients with DM demonstrates lower excitability than the control adult isoform [18].
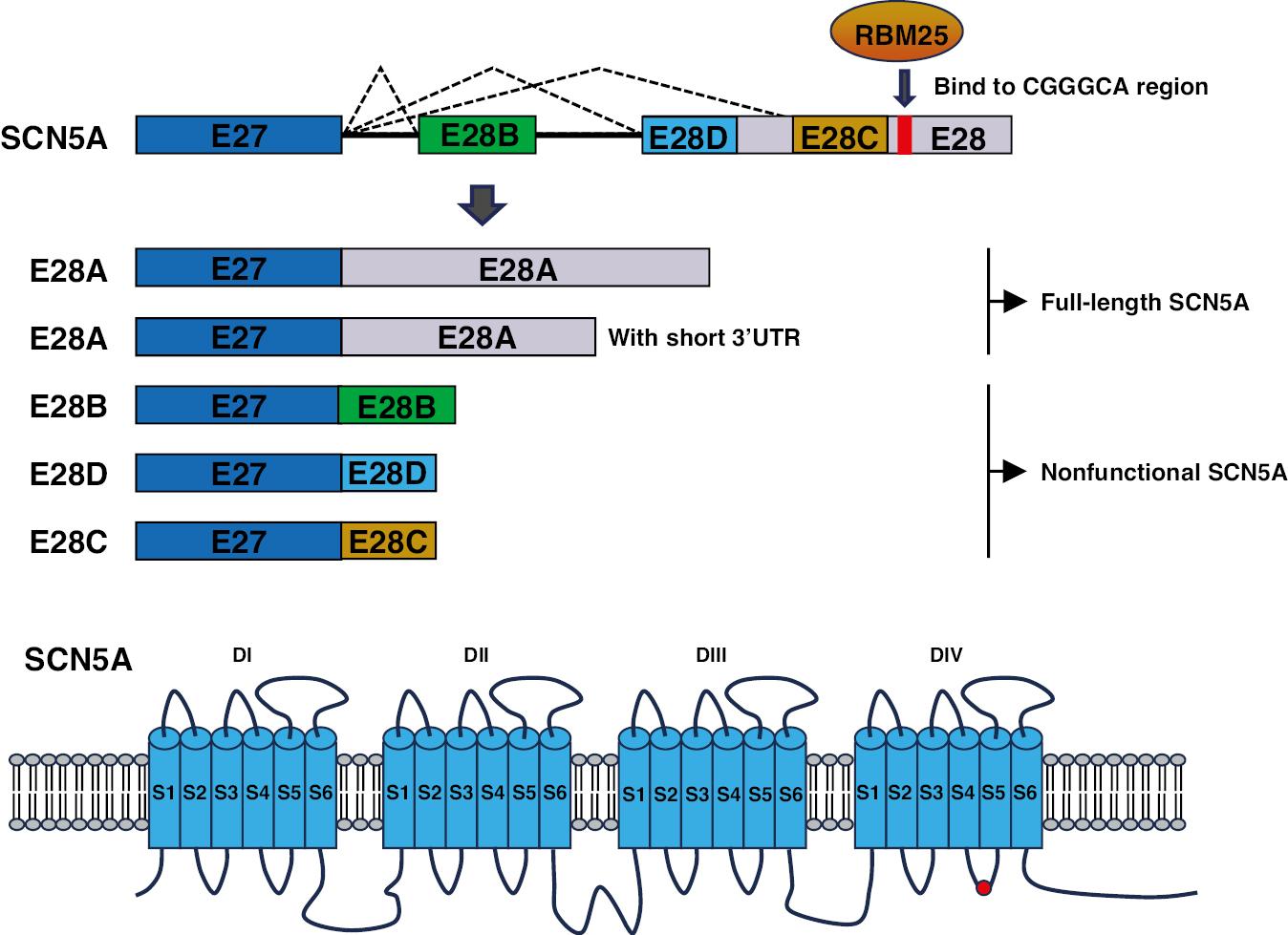
Diagram Illustrating Alternative Splicing of the SCN5A.
Top: Partial representation of the SCN5A genomic/mRNA sequence, focusing on selected exons rather than the full set of 16 exons. Bottom: The SCN5A protein comprises four homologous transmembrane domains (DI–DIV), each containing six transmembrane segments (S1–S6). The S1–S4 segments in each domain constitute the voltage-sensing regions, whereas the S5–S6 segments, along with the intervening loop, form the central pore and selective filter of the channel. (Red rectangle and red circle: RBM25 binding site.)
The KCNQ1 gene encodes a protein that belongs to the voltage-gated potassium channel family, which is crucial in regulating potassium ion flow across cell membranes. Mutations in the KCNQ1 gene are associated with various cardiac disorders, notably LQTS. Specifically, mutations in exon 7 result in exon-skipping events in KCNQ1 mRNAs and consequently the LQT1 phenotype [44]. These AS variants exert dominant-negative effects by sequestering wild-type proteins intracellularly, thereby impeding their translocation to the plasma membrane [45]. Additionally, nine isoforms of the voltage gated sodium channel NaV1.5 have been identified. Multiple NaV1.5 isoforms are functionally expressed in the brain and display distinct expression patterns with respect to cardiac NaV1.5 [46].
Alternative Splicing and Signaling Pathways
Insulin-like growth factor (IGF-1) plays crucial roles in various physiological functions through the production of multiple isoforms via AS. The diversity of IGF-1 is achieved at the N-terminal end of the protein through two alternative promoters, which initiate transcription from either exon 1 or exon 2, and at the C-terminal end by AS of exons 5 and 6, thus forming isoforms classified as A, B, or C (Figure 6). These distinct isoforms interact with the extracellular matrix with varying affinities and consequently influence IGF-1 bioavailability. Notably, IGF-1 isoform A promotes vessel formation after injury, and facilitates crosstalk between the bone marrow and heart through the release of specific cytokines [47]. Furthermore, Multiple IGF-1 propeptides produced by alternative exon splicing are cleaved to yield a common 70-amino acid core hormone, which is released into the bloodstream. This 70-amino acid core hormone attenuates oxidative stress and enhances cardiac function [48].
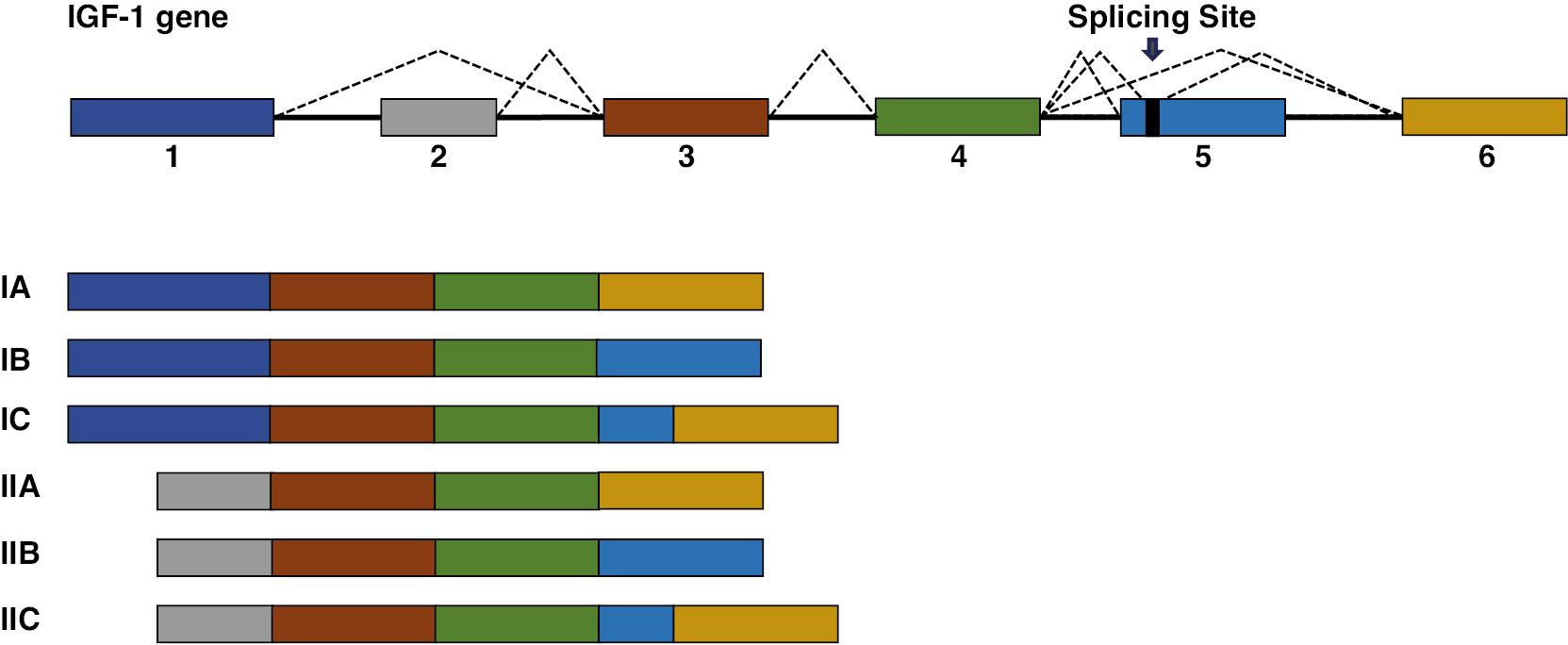
Schematic Representation of the IGF-1 Gene Structure and its Alternatively Spliced Isoforms.
The IGF-1 gene consists of six exons. Alternative splicing of exons 1 and 2 determines whether the transcript belongs to IGF-1 class I or II. Exons 3 and 4 are present in all known isoforms. The inclusion or exclusion of exons 5 and 6 gives rise to the different isoforms, classified as A, B, or C.
The vascular endothelial growth factor (VEGF) family of proteins regulates blood flow, growth, and function in both normal physiology and disease processes. AS of VEGF-A yields multiple isoforms within two subfamilies, each with specific functions. AS of exons 5–7 of the VEGF gene generates forms with differing bioavailability and activities. Additionally, alternative splice-site selection in exon 8 generates either proangiogenic proteins, termed VEGFxxx, or antiangiogenic proteins, termed VEGFxxxb [49]. The VEGF family isoforms, represented as VEGFxxx, where “xxx” denotes the specific amino acid sequence length, are produced through differential splicing of exons 6 and 7, as well as the proximal splice site in exon 8 (known as exon 8a). This splicing process generates two distinct families of proteins, VEGFxxx and VEGFxxxb, each differing by just six amino acids at their C-terminus. The VEGFxxx family is generated through proximal splice-site selection in exon 8, which produces proangiogenic isoforms. In contrast, distal splice-site selection, occurring 66 base pairs downstream, results in the formation of the antiangiogenic family, VEGFxxxb. This distinction in splicing not only affects the amino acid composition but also influences their functional roles in angiogenesis.
The X-box binding protein 1 (XBP1), a stress-inducible transcription factor found in both invertebrates and vertebrates, plays crucial roles in cell survival during stress conditions. In mammalian cells, XBP1 is a critical signal transducer in the endoplasmic reticulum (ER) stress response. Additionally, XBP1 mRNA splicing is intricately associated with VEGF signaling, and contributes to endothelial cell proliferation and angiogenesis in ischemic tissues [50]. Moreover, XBP1 directly binds the promoter region and modulates the expression of brain natriuretic peptide (BNP) in cardiomyocytes [51].
Myocardin functions as a transcriptional co-activator of serum response factor (SRF), and plays a critical role in regulating the expression of genes specific to cardiac and smooth muscle tissues. Through AS, myocardin generates a cardiac-enriched isoform with a distinct peptide sequence enabling it to interact with and enhance the transcriptional activity of MEF2 [52].
In the heart, AS generates two isoforms of TBX5. The longer isoform (TBX5a) is a primary regulator of cardiac lineage specification and heart development. AS-mediated inclusion of exon 8 in TBX5 introduces a premature stop codon leading to the production of a truncated protein devoid of transcription activity (TBC5b) [53]. Additionally, TBX5 interacts with the trans-regulatory splicing factor SRSF2 in modulating AS [21].
Alternative Splicing and Splicing Factors
RBPs are essential regulators within cells that govern processes such as mRNA transport, splicing, editing, and stability. Two extensively studied RBP families are the SR protein family and the hnRNP family. These proteins are widely expressed across cell types and play critical roles in spliceosome assembly. SR proteins typically feature at least one RRM and an RS domain. The RRM domain facilitates RNA binding, whereas the RS domain serves as a hub for protein interactions. The hnRNP family is named for its association with heterogeneous nuclear RNAs, also known as pre-mRNAs. Members of this family typically contain an RNA-binding motif alongside other domains governing protein-protein interactions or cellular localization. Over time, the boundaries between the SR and hnRNP families have become less distinct, because of their overlapping properties and functions. Beyond these broadly expressed RBPs, numerous tissue-specific or enriched RBPs exist. Whereas some RBPs within the SR and hnRNP families might appear redundant, the specific combination of RBPs, including tissue-specific variants, within the spliceosome, determines their ability to recognize alternative exons (Table 2).
Summary of RBPs in Cardiac Disease.
| RBPs | Example genes regulated by RBPs | AS type | Disease | Reference |
|---|---|---|---|---|
| RBM20 | TTN | IR | DCM | [4] |
| RBFox1 | Cav1.2 | ES | DCM | [54] |
| RBM24 | TTN | IR | DCM | [55] |
| RBM25 | SCN5A | ES | HF | [43] |
| RBPMS | TTN, Pdlim5, Nexn | ES, IR | DCM, HF | [1] |
| SRSF1 | VEGF | A3SS | MI | [56] |
| SRSF3 | mTOR | A3SS | HF | [57] |
| SRSF5 | Myom1 | IR | NVM | [58] |
| SRSF10 | MEF2A, CASQ2 | ES | DCM | [35] |
| hnRNPA1 | Pkm1/Pkm2 | MXE | IHD | [59] |
| hnRNPU | Camk2d, TTN | IR | Cardiogenesis | [60] |
| hnRNPC | YAP1 | ES | HF | [61] |
| SF3B1 | KHK | MXE | HF | [62] |
| MBNL | SCN5A | MXE | DM | [18] |
| CELF1 | Ablim3, Per1, Gpr116 | ES | T1DM | [63] |
| CELF4 | TNNT | IR | Cardiogenesis | [64] |
| QKL | Tpm1, TTN | ES, IR | HF | [65] |
| LUC7L3 | SCN5A | ES | HF | [43] |
| SLM2 | TTN | ES | HF | [66] |
Note: ES, exon skipping; MXE, mutually exclusive exon usage; A5SS/A3SS, alternative 5′SS/3′SS selection; IR, intron retention; IHD, ischemic heart disease; T1DM, type1 diabetes mellitus; NVM, noncompaction of ventricular myocardium.
RBM20 functions as a splicing factor that targets several critical cardiac genes, including TTN and CAMK2D. Dysregulated TTN splicing is believed to be the primary driver of RBM20-induced DCM. Additionally, RBM20 plays a role in the AS of calcium and ion-handling genes, notably CaMKIIδ and RyR2 [67]. Mutations in RBM20 have been associated with 2–3% of all idiopathic DCM cases and as many as 13% of cases involving sudden cardiac death. RBM20 binds a cluster containing most RBM20 binding motifs via its RNA recognition motif domain, and represses both upstream and downstream introns. The regulation of exon exclusion involves PTB4, a novel TTN splice regulator counteracting the repressive activity of RBM20. The regulatory actions of RBM20 and PTB4 contribute to the modulation of TTN elasticity in cardiac disease [68]. Notably, a family affected by DCM has been found to carry an E913K mutation within a glutamate-rich region of RBM20; this mutation leads to RBM20 haploinsufficiency, disruption of AS of TTN, and ultimately a significant shift toward highly compliant titin isoforms and impairment of the Frank-Starling mechanism [4].
RBM24 is a critical regulator of AS events involved in the development and pathology of cardiac and skeletal muscle tissues. Its interaction with an ISE is both necessary and sufficient to counteract the repression of exon inclusion exerted by an ESS, which contains PTB and hnRNP A1/A2 binding sites [69]. RBM24 regulates AS of key genes involved in myofibrilla genesis, including ACTN2, TTN, and MYH10. Notably, the RBM24-mediated inclusion of exon 6 in ACTN2 during distinct stages of cardiac differentiation plays crucial roles in ensuring proper sarcomere assembly and maintenance of sarcomeric integrity [70]. Additionally, aberrant SCN5A mRNA splicing mediated by RBM25/LUC7L3 decreases the Na+ channel current in the context of HF [43].
RNA binding protein with multiple splicing (RBPMS) plays a critical role in cardiac development by modulating mRNA splicing. Loss of RBPMS specifically in cardiac tissue leads to severe contractile defects in cardiomyocytes, and ultimately DCM and early lethality in adult mice. RNA sequencing studies have revealed that RBPMS regulates the AS of mRNA transcripts encoding proteins essential for sarcomere structure and function, including TTN, Pdlim5, and Nexn, thereby generating novel protein isoforms [1].
RBFox1 and RBFox2, key players in AS, are important in cardiac transcriptional reprogramming and notably influence the pathogenesis of HF. Zhou et al. have demonstrated that RBFox2 dynamically regulates the function of vascular CaV1.2 calcium channels by modulating the expression of alternative exons 9* and 33, thereby affecting vascular myogenic tone [71]. Moreover, He et al. have identified a splice variant of the CaV1.2 channel in rodents, termed CaV1.2e21+22, which includes a pair of mutually exclusive exons, exons 21 and 22. This variant decreases the cell-surface expression of wild-type CaV1.2 channels and consequently whole-cell Ca2+ influx via CaV1.2 channels [72]. Zebrafish lacking two orthologs of the RNA binding protein RBFox2 exhibit cardiovascular abnormalities resembling those observed in patients with hypoplastic left heart syndrome, including defects in ventricular, valve, and aortic development. Mechanistically, the loss of RBFox2 decreases the expression and AS of sarcomere and mitochondrial components, thus compromising sarcomere assembly and mitochondrial respiration, respectively [73].
SRSF1 orchestrates the AS of CaMKIIδ, thereby temporally reprogramming excitation-contraction coupling in cardiac muscle [16]. SRSF10 regulates cardiac-specific AS of triadin pre-mRNA and plays crucial roles in proper Ca2+ handling during embryonic heart development [74]. SRSF5 promotes the AS of Myom1 (myomesin-1), and its knockout in mice leads to cardiac dysfunction characterized by noncompaction of the ventricular myocardium [58]. Additionally, Ortiz-Sanchez et al. have reported that cardiomyocyte-specific loss of SRSF3 expression results in widespread changes in AS and decapping of critical mRNAs involved in cardiac contraction. This effect is mediated by the generation of a short mTOR isoform through AS, which in turn decreases 4E-BP1 phosphorylation [57].
hnRNPA1 plays crucial roles in embryonic muscle development by modulating the expression and AS of genes associated with muscle function [75]. hnRNPU is essential for heart development and function, by exerting regulatory control over AS processes. Loss of hnRNPU expression in cardiomyocytes results in aberrant splicing of Junctin pre-mRNA. The protein product of an alternatively spliced Junctin isoform is N-glycosylated at a specific asparagine site. The asparagine site is required for interactions with specific protein partners [60].
hnRNPC is up-regulated and translocates to the sarcomeric Z-disc during extracellular matrix pathological remodeling. Changes in hnRNPC expression, phosphorylation, and subcellular localization are mechanistically linked and affect the AS of mRNAs implicated in mechanotransduction and CVD. Notably, these alterations influence the Hippo pathway effector Yes-associated protein 1 [61]. SF3B1 is associated with pathological cardiac hypertrophy. Activation of SF3B1 by HIF1α results in splice switching of KHK-A to KHK-C, thereby modulating the activity of the central fructose-metabolizing enzyme [62].
The CELF family proteins act as splicing factors that mediate AS by binding the CUG sequence in pre-mRNAs. The single-nucleotide polymorphism rs1786814 on the CELF4 gene shows a dose-dependent association with anthracycline-induced cardiomyopathy [76]. Repression of CELF proteins in mouse hearts disrupts the AS of genes associated with cardiogenesis, such as Mef2a, and contributes to a phenotype resembling cardiomyopathy [77]. MBNL proteins play critical roles in regulating AS. Deficiency in MBNL1/2 contributes to a portion of the mis-splicing events observed in the hearts of individuals with myotonic dystrophy type 1. Mice lacking both MBNL1 and MBNL2 demonstrate sudden cardiac death and exhibit molecular signatures reminiscent of myotonic dystrophy [78].
QKI, a crucial regulator of AS in both human and mouse cardiomyocytes, plays indispensable roles in cardiogenesis and heart function [79]. Adult mice with tamoxifen-inducible QKI deletion rapidly develop HF with severe disruption of sarcomeres. RNA sequencing studies have identified QKI as a regulator of AS in more than 1000 genes, including those encoding sarcomere and cytoskeletal components, calcium-handling proteins, and transcriptional regulators [65].
SLM2, a newly characterized splicing factor implicated in the development of DCM, is upregulated in the hearts of patients with DCM, and binds critical transcripts encoding sarcomere constituents such as MYL2, TNNI3, TNNT2, TPM1/2, and TTN. Mechanistically, SLM2 facilitates intron retention and inhibits exon exclusion, thereby mediating the AS of mRNA regions encoding the variable proline-, glutamate-, valine-, and lysine-rich (PEVK) domain, and another portion of the I-band region of titin [80]. hnRNPL (PTBP1) finely modulates the sensitivity of CaV1.2 channels to dihydropyridine by regulating the utilization of exon 8/8a and leading to alterations in dihydropyridine-induced vasodilation responses [81].
Alternative Splicing of Long Non-coding RNAs
Mounting recent evidence suggests that non-coding RNAs play crucial roles in the cardiovascular system. Long non-coding RNAs (lncRNAs) are transcripts longer than 200 nucleotides that are distinguished from microRNAs and protein-coding transcripts in that they do not encode proteins. Because of their tissue- or cell-type specific expression and potential regulatory roles, lncRNAs are assumed to have important functions during organismal development, including in the heart.
The lncRNA lncMYH7b is generated through AS of the Myosin heavy chain 7b (MYH7b) gene in mammalian cardiac muscle. Unlike the MYH7b gene, which does not produce a full-length protein, lncMYH7b is formed through post-transcriptional exon-skipping splicing. lncMYH7b plays a regulatory role in modulating the ratio of MYH7 to MYH6, a critical determinant of cardiac contractility. Although the exact regulatory mechanisms of lncMYH7b on MYH7 and MYH6 ratios have not been fully elucidated, lncMYH7b has been hypothesized to exert its regulatory function through interaction with TEAD and MEF2 family transcription factors. These factors play key roles in cardiac gene regulation, and might be involved in modulating the expression of MYH7 and MYH6 isoforms. Additionally, lncMYH7b is involved in regulating beat rate and sarcomere formation in cardiomyocytes [82].
Mhrt lncRNAs are generated through AS of MYH7 antisense transcripts incorporating partial sequences of MYH7 introns and exons. Mhrt transcripts are highly abundant and specifically expressed in the adult heart. Pathological stress triggers the activation of the Brg1-Hdac-Parp chromatin repressor complex and leads to the suppression of Mhrt transcription in the heart. The stress-induced repression of Mhrt serves as a protective mechanism that restores Mhrt levels to pre-stress conditions and safeguards the heart against hypertrophy and failure [83].
The lncRNA CARMEN generates a range of alternatively spliced isoforms, which play a critical role in dictating cell fate specification toward cardiomyocyte and SMC lineages. One such isoform, CARMEN-201, is encoded within an alternatively spliced exon containing an MIRc short interspersed nuclear element. This element binds the transcriptional repressor REST and directs it to cardiogenic loci, such as ISL1, IRX1, IRX5, and SFRP1, thereby inhibiting the CM gene program. Consequently, the expression of genes involved in SMC differentiation is induced [84]. Although a direct association has not been demonstrated between lncCARMEN and a specific CARMEN protein, lncCARMEN has been implicated in the modulation of gene expression networks involved in cardiac lineage commitment. Manavski et al. have identified that the lncRNA Snhg15 is downregulated in the cardiac tissue in patients with HF. Snhg15 directly interacts with ZRANB2, an important regulator of AS [26].
The lncRNA lncCM1 is regulated during aging in cardiomyocytes and has been hypothesized to exert a protective effect on the myocardium. lncCM1 regulates AS and functions as an anti-apoptotic lncRNA [27]. MALAT1 interacts with SR proteins, and influences the distribution of these and other splicing factors within nuclear speckle domains. Depletion of MALAT1 or overexpression of an SR protein alters the AS of a similar set of endogenous pre-mRNAs. Additionally, MALAT1 regulates the cellular levels of phosphorylated forms of SR proteins [85].
Alternative Splicing Therapy
Antisense Oligonucleotides
AS can be modulated with small modified antisense oligonucleotides (AONs). These oligonucleotides contain an antisense sequence that binds a splicing regulatory element within the target exon, thereby disrupting splicing and preventing the exon’s inclusion in the final transcript. AONs are chemically modified at their backbones to increase stability, enhance target selectivity, and protect RNA-RNA interactions against degradation by RNase H. Exon skipping facilitated by AONs is a promising therapeutic strategy for AS-related disorders.
A mouse model of hypertrophic cardiomyopathy contains a Mybpc3-targeted knock-in with a homozygous G>A transition in exon 6, thus resulting in the production of three distinct aberrant mRNAs. To counteract these aberrant mRNAs, AON-5 and AON-6 have been designed to mask splicing enhancer motifs in exons 5 and 6, respectively. Transduction of knock-in mice with AON-5 and AON-6 decreases aberrant mRNAs, alleviates cardiac dysfunction, and prevents left ventricular hypertrophy [86].
Myotonic dystrophy type 1, a genetic disease and the most prevalent form of muscular dystrophy in adults, is characterized by abnormal expansion of CTG repeat sequences in the dystrophia myotonica-protein kinase (DMPK) gene. This expanded CTG repeat leads to sequestration and functional loss of the splicing factor MBNL1 in the cell nucleus, and consequently contributes to the pathogenesis of DM1. An AON has been developed to target CTG repeats, thereby disrupting the interaction with MBNL1. Liberation of MBNL1 from sequestration restores its function in AS regulation and ameliorates myotonic dystrophy [87].
A similar therapeutic strategy has been used to address Duchenne muscular dystrophy (DMD), a condition characterized by various mutations in the dystrophin gene that lead to premature protein truncation. By using AONs to induce the skipping of specific exons, the open reading frame can be restored, thus resulting in the synthesis of a partially functional protein. DMD is typically caused by mutations in the dystrophin gene that disrupt the open reading frame, whereas the milder Becker muscular dystrophy results from mutations that preserve an in-frame mRNA transcript, and allow for the production of a protein with intact N- and C-terminal functional domains. Studies have shown that excluding specific exons affected by frame-shifting mutations can restore the open reading frame and result in the production of a partially functional protein [88]. This targeted exon skipping is achieved through the administration of oligonucleotides complementary to sequences crucial for the normal splicing of the exon into the transcript (Figure 7A). In 2018, the FDA approved oligonucleotide agents aimed at excluding exon 51 for DMD treatment.
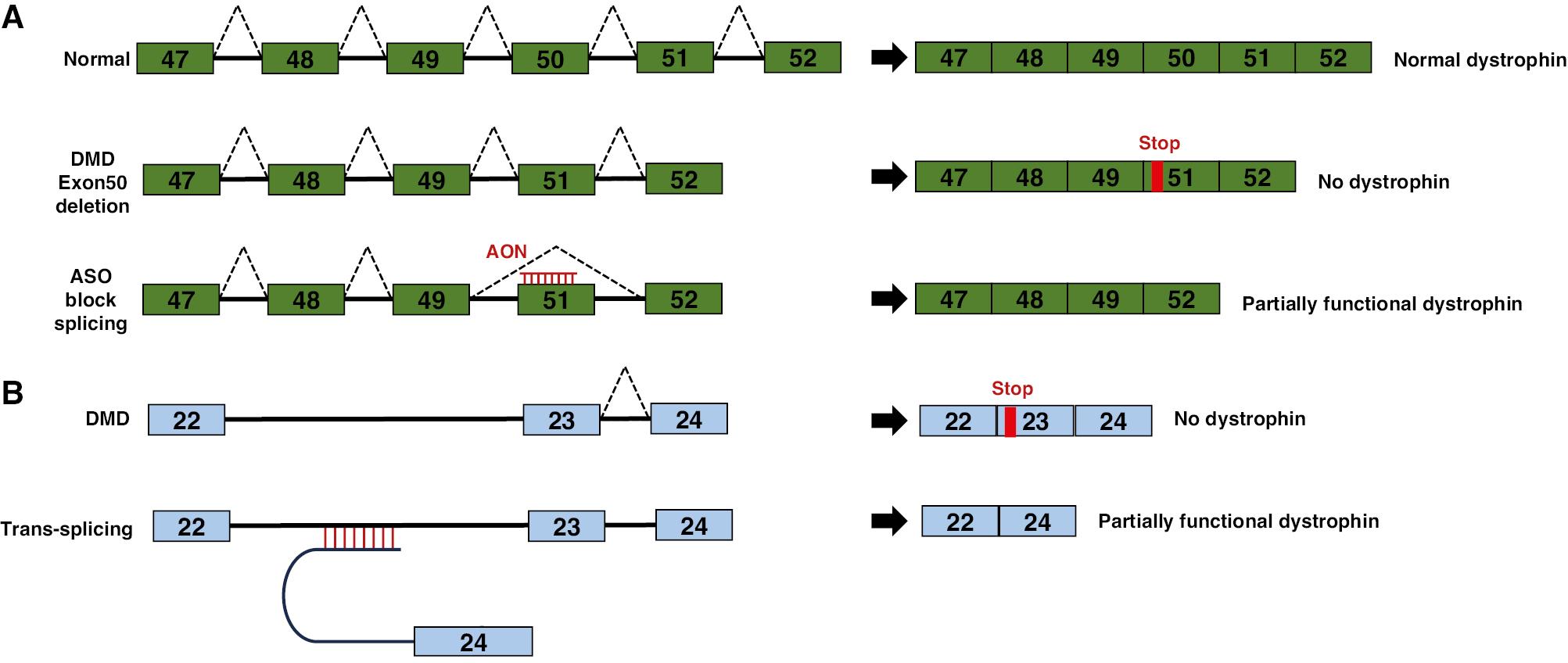
Reprogramming of mRNA Splicing with Antisense Oligonucleotides (AONs).
(A) Exon skipping to restore dystrophin production in Duchenne muscular dystrophy (DMD). Under normal conditions, splicing of DMD pre-mRNA results in a mature mRNA containing all exons required for the synthesis of full-length dystrophin. In DMD, mutations in exon 50 lead to abnormal splicing excluding exon 50 from the mature mRNA, thus causing a frameshift and loss of dystrophin production. AON binding to exon 51 disrupts its splicing signal and causes exon 51 to be skipped along with exon 50. Subsequently, the original reading frame is restored, and a shortened, but partially functional, dystrophin protein is produced. (B) The murine dystrophin minigene consists of exons 22 through 24, with their respective introns (depicted as lines). A mutation in exon 23 is shown in red. The trans-splicing molecule AS-E24 includes a 150-nucleotide antisense sequence complementary to intron 22, along with a spacer, a conserved yeast branch point sequence, a polypyrimidine tract, a 3′ acceptor site, and exon 24.
Additional studies have explored the conjugation of an arginine-rich cell-penetrating peptide to the oligonucleotide, thus resulting in Pip5e-PMO. The hydrophobic core of the Pip sequences aids in delivering the oligonucleotide to the heart, and specific modifications can further enhance its activity in cardiac tissues [89]. Another approach has used an adeno-associated virus vector for expression of AONs associated with a modified U7 small nuclear RNA to achieve sustained exon skipping and remove the mutated exon from the dystrophin messenger mRNA [90].
Trans-splicing
RNA trans-splicing has emerged as a potential therapeutic approach for various genetic diseases, including hypertrophic cardiomyopathy caused by mutations in the Mybpc3 gene. These mutations can be corrected via RNA trans-splicing to produce normal Mybpc3 proteins. Trans-splicing is induced between two independently transcribed molecules: the mutant endogenous Mybpc3 pre-mRNA and an engineered pre-trans-splicing molecule (PTM) carrying a FLAG-tagged wild-type Mybpc3 cDNA sequence. PTMs are delivered via adeno-associated virus transduction of cultured cardiac myocytes [91]. The repaired cMyBP-C protein has been detected by immunoprecipitation both in vitro and in vivo, and demonstrated to be correctly incorporated into the sarcomeres of cardiac myocytes. Furthermore, Mybpc3 trans-splicing occurs in human induced pluripotent stem cell-derived cardiomyocytes, thereby restoring cMyBP-C levels to 81% of control levels. This process suppresses hypertrophy and partially restores gene expression to control levels in hypertrophic cardiomyopathy cells [92]. The trans-splicing system is designed to rescue mutations in the dystrophin gene. In a mouse model carrying a nonsense mutation in exon 23 of the dystrophin gene, researchers have tested the feasibility of double trans-splicing as a therapeutic strategy. To do so, they constructed a minigene containing a murine dystrophin genomic fragment spanning from exon 22 to exon 24, which included the mutated exon 23, with expression driven by a constitutive promoter. The trans-splicing molecule AS-E24 includes a 150-nucleotide antisense sequence complementary to intron 22, along with a spacer, a conserved yeast branch point sequence, a polypyrimidine tract, a 3′ acceptor site, and exon 24. This minigene was then transfected into a mouse model. The results demonstrated precise and efficient replacement of exon 23 in the target mRNA, thus confirming the potential of this approach (Figure 7B).
Potential and Limitations of Alternative Splicing Events as Biomarkers for Cardiac Diseases
AS events have substantial potential as biomarkers for cardiac diseases yet also present notable limitations. One benefit is that AS events can reflect disease-specific patterns, thus enabling early diagnosis and intervention. For instance, distinct splicing variants associated with conditions such as HF or hypertrophic cardiomyopathy may serve as indicators of disease onset and progression, and therefore facilitate timely treatment strategies. Furthermore, AS biomarkers could contribute to personalized medicine by allowing for tailored therapies based on an individual’s unique splicing profile.
However, several limitations must also be addressed. The complexity and variability of AS across different tissues and developmental stages complicate the interpretation of AS patterns as reliable biomarkers. Additionally, technical challenges in detecting low-abundance splicing variants in non-invasive samples hinder their clinical applicability. Not all AS events are functionally relevant to disease, and distinguishing those that contribute to pathology from those that are incidental remains challenging. Finally, the translation of AS biomarkers into clinical practice requires standardized assays and regulatory considerations. Overcoming these challenges will be crucial for effectively leveraging AS events in precision cardiology.
Discussion
The roles of individual splicing isoforms in heart disease have garnered increasing attention and understanding over time. Dysregulation of splicing factors such as RBPs, along with their targets associated with sarcomere function, ion channels, and lncRNAs, are well-documented in the context of heart disease. These splicing factors are critical in maintaining normal cardiac function, and their disruption can lead to aberrant splicing events and ultimately contribute to disease progression. For example, splicing factors such as SRSF2 and RBM20 have been shown to regulate essential genes involved in cardiac contraction and ion homeostasis. The misregulation of these splicing factors alters the expression of key isoforms, and consequently can disrupt the normal physiology of the heart and lead to pathological states, such as arrhythmias and cardiomyopathies.
Despite these advancements, a substantial gap in persists understanding of the regulation of global AS patterns in heart disease. Although specific splicing factors and isoforms have been identified, a broader understanding of how AS is regulated at the transcriptome level remains lacking. The integration of next-generation sequencing techniques, such as RNA-Seq, has greatly enhanced the ability to understand transcriptome-wide alterations. These technologies have enabled previously unknown splicing variants to be uncovered and overarching splicing patterns associated with CVD to be identified. For instance, next-generation sequencing has revealed how certain splicing events, such as exon skipping or the use of alternative polyadenylation sites, contribute to the development of heart disease by altering the functions of key genes.
In recent years, therapeutic strategies targeting AS have rapidly evolved, and currently offer promising avenues for the treatment of muscular and CVD. AONs, for example, have been developed to modulate specific splicing events by promoting the inclusion or exclusion of particular exons. These strategies are already being applied in clinical settings for muscular dystrophies, and their potential for application in heart disease is growing. Furthermore, efforts to target specific splicing isoforms for therapeutic intervention are showing promising outcomes. For example, modulating the splicing of the RBM20 gene has been proposed as a potential treatment for DCM, thus highlighting the therapeutic potential of splicing modulation in CVD.
However, an urgent need exists to identify novel splicing regulators and critical pathological AS targets in heart disease. The discovery of new splicing factors or regulatory mechanisms might unlock further therapeutic possibilities. Additionally, understanding how these regulators contribute to disease-specific splicing patterns could provide deeper insights into the molecular underpinnings of CVD. For instance, isoforms that might be harmless under normal conditions could become pathological under stress or during aging, thus further complicating the splicing landscape in heart disease.
In conclusion, although substantial progress has been made in understanding the roles of splicing factors and specific splicing isoforms in heart disease, much remains to be explored. The integration of cutting-edge sequencing technologies and the development of splicing-modulating therapies have great promise for advancing understanding and treatment of CVD. Future research should focus on uncovering novel splicing regulators and identifying critical splicing events that might serve as therapeutic targets, thereby providing new avenues for the treatment of CVD.
