
Heart Failure Classification, Epidemiology, and Prognosis
Heart failure (HF), a clinical syndrome caused by a variety of disturbances in cardiac structure and/or function, leads to ventricular systolic and/or diastolic dysfunction and insufficient delivery of blood and oxygen to the surrounding tissues to fulfill their metabolic needs [1]. This complex clinical syndrome is characterized by pulmonary or systemic congestion. Currently, HF can be classified into four types according to left ventricular ejection fraction (LVEF): HF with decreased ejection fraction (HFrEF; LVEF ≤40%), HF with preserved ejection fraction (HFpEF, LVEF ≥50%), HF with mildly decreased EF (HFmrEF, LVEF 40–49%), and HF with improved EF (HFimpEF), with respect to a baseline LVEF of ≤40%, a ≥10% increase from baseline LVEF, and a second measurement of LVEF of >40% [2].
HF affects more than 64 million people worldwide [3]. The China Hypertension survey, enrolling 22,158 participants, has revealed a weighted prevalence of HF among the Chinese population ≥35 years of age of 1.3% (estimated 13.7 million) [4]. Of these, 1.4% of participants exhibited HFrEF, and 2.7% were classified as having moderate or severe diastolic dysfunction. In the United States, the prevalence of HF was 2.42% in 2012 and is expected to rise to 2.97% by 2030 [5]. In European countries, the prevalence of HF has been estimated to be 17.20 cases per 1000 people [6]. In Australia, the HF prevalence ranges between 1.0% and 2.0% [7]. Most reports on the epidemiology of HF have been conducted in developed countries, whereas reliable estimates of the prevalence of HF in middle- and low-income countries are lacking. However, HF is clearly the fastest-growing cardiovascular disease worldwide. The prevalence of HFpEF is increasing and currently exceeds that of HFrEF, whereas the prevalence of HFrEF appears to be stable or even declining. This trend may largely account for the primary prevention of cardiovascular disease and improved management of ischemic heart disease. In contrast, the risk factors for HFpEF, such as aging, obesity, diabetes, and hypertension, have markedly increased in this century [8].
In recent years, with medical developments, the 5-year HF survival rate in developed countries is substantially greater than that 20 years ago. A 2016 study of HF in the UK has indicated that the overall 1-year, 5-year, and 10-year survival rates of patients with HF increased by 6.6% (from 74.2% in 2000 to 80.8% in 2016), 7.2% (from 41.0% in 2001 to 48.2% in 2012), and 6.4% (from 19.8% in 2005 to 26.2% in 2007), respectively [9]. Similarly, the 5-year HF survival rate was 48% in Sweden [9]. The China Heart Failure Registration Research (China-HF) analysis of 13,687 patients with HF in 132 hospitals across the country from January 2012 to September 2015 has revealed a 4.1% fatality rate among hospitalized patients with HF [10]. According to the 2020 China Heart Failure Medical Quality Control Report, an analysis of 33,413 patients with HF admitted to 113 hospitals across the country between January 2017 and October 2020 has revealed an in-hospital mortality rate of 2.8% [11]. Despite notable advancements in the treatment and prevention of HF in recent decades, the 5-year HF survival rate has improved; however, the overall prognosis remains poor, and further research is required.
Inflammatory Biomarkers in Heart Failure
Systemic inflammation is intimately involved in the pathophysiology of HF [12]. In 1990, circulating levels of tumor necrosis factor were observed to be elevated in patients with HF, thus providing the earliest evidence of sustained inflammation in HF [13]. Subsequent experimental and clinical findings have highlighted that the activation of immune systems aggravates cardiac dysfunction, and targeting inflammatory responses is a potential therapeutic strategy for HF. Consequently, inflammatory biomarkers might serve as potential targets to regulate immune systems in HF (Figure 1).
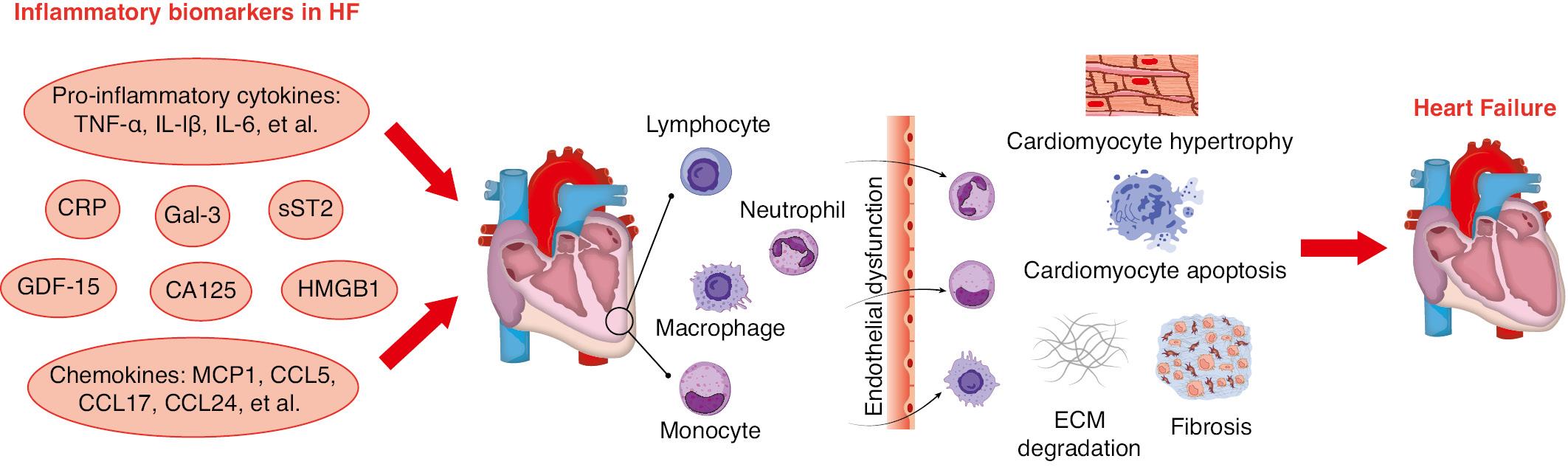
The inflammatory biomarkers in HF.
The circulating levels of inflammatory biomarkers such as pro-inflammatory cytokines (TNFα, IL-1β, IL-6, et al), chemokines (MCP1, CCL5, CCL17, CCL24, et al.), soluble tumor suppressant factor-2 (sST2), growth differentiation factor-15 (GDF-15), high mobility group 1 (HMGB1), carbohydrate antigen 125 (CA125), C-reactive protein (CRP), and galectin-3 (Gal-3) have been shown to be significantly elevated in patients with HF. These inflammatory biomarkers play an important role in the development of HF via promoting leukocyte migration and infiltration, triggering endothelial dysfunction and apoptosis of cardiomyocyte, activating matrix metalloproteinases and degrading extracellular matrix, and eliciting cardiac hypertrophy and fibrosis.
Pro-Inflammatory Cytokines
Cytokines are low-molecular-weight bioactive proteins that regulate cell function in an autocrine or paracrine manner [14]. Elevated circulating pro-inflammatory cytokine concentrations in patients with HFrEF or HFpEF are a well-known characteristic associated with poorer clinical outcomes [15–17]. The most important pro-inflammatory cytokines involved in HF include tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), and IL-6. Experimental models have shown that pro-inflammatory cytokines may produce negative inotropic effects [18], and induce activation of matrix metalloproteinases (MMPs) that disturb the structure of the myocardial extracellular matrix [19]; induce hypertrophy and promote cardiomyocyte apoptosis [20]; and stimulate a fibrogenic program, induce the production of fibroblast growth factors, and eventually lead to myocardial fibrosis [21]. Cardiac-specific overexpression of TNF in mice can activate MMPs, thus producing a classic spontaneous HF phenotype [19]. MMPs, a family of proteases, degrade extracellular matrix proteins and determine the interstitial architecture. An imbalance between the activity of MMPs and tissue inhibitor of MMPs (TIMPs) is associated with the progression of HF [22]. Moreover, an increase in the ratio of MMPs/TIMPs leads to degradation of the extracellular matrix, whereas a decrease in this ratio leads to progressive myocardial fibrosis [23]. Therefore, cytokines may also elicit a direct effect on the pathogenesis of HF by regulating the expression of both MMPs and TIMPs.
Chemokines
Chemokines are small glycoproteins that stimulate leukocyte migration and infiltration, and induce the production of pro-inflammatory cytokines and reactive oxygen species [24]. The four known chemokine subfamilies, distinguished by their primary amino acid sequences, are CXC, CC, C, and CX3C [25]. Patients with HF exhibit significantly higher plasma chemokine concentrations than healthy individuals, including CC-chemokine ligand 2 (CCL2; also known as MCP1) [26], CCL3 (also known as MIP1α) [27], CCL5 (also known as RANTES) [28], CCL17 [29], CCL21 [30], and CCL24 [31]. Animal experiments have revealed that anti-CCL5 mAb decreased leucocyte recruitment, infarct size, and improved the post-infarction HF [32]. CCL17 knockout or anti-CCL17 neutralizing antibody significantly repressed cardiac hypertrophy and fibrosis by reducing the T helper 17 (Th17)/regulatory T cell (Treg) ratio and promoting Th1-type polarization, in a mouse model of age-related and angiotensin II–induced HF [31]. Therefore, the level of circulating chemokines can be considered to indicate the severity and prognosis of HF, and may be a potential intervention target.
C-Reactive Protein
C-reactive protein (CRP), produced by the liver during inflammation, is widely used to assess systemic inflammation. A prospective cohort study has suggested that elevated CRP is an independent risk marker and is associated with poor prognosis for HF [33]. Similarly, a meta-analysis of 19 studies has revealed that CRP can serve as a biomarker to predict the development of HFpEF and long-term clinical outcomes in patients with HFpEF [34]. Notably, for every 1 mg/L increase in CRP, the risk of HF increases by 10%, and the risk of HFrEF and HFpEF increases by 9% and 12%, respectively [33]. Therefore, CRP might serve as an important inflammatory response marker reflecting HF severity and prognosis.
Growth Differentiation Factor-15
Growth differentiation factor-15 (GDF-15), an inflammatory stress marker, is a member of the transforming growth factor-beta superfamily. GDF-15 has a protective effect on the ischemic myocardium, and GDF-15 deficiency causes loss of anti-inflammatory mechanisms in mice that can lead to fatal cardiac rupture after myocardial infarction [35]. A clinical study has revealed that GDF-15 is associated with long-term mortality in patients with acute HF and can predict the prognosis of patients with acute HF independently of BNP [36]. Therefore, GDF-15 may serve as a new indicator for diagnosis and follow-up of patients with HF and may be used as an anti-inflammatory therapy target for HF in the future.
Other Inflammatory Biomarkers in Heart Failure
High mobility group box 1 (HMGB1), a non-histone DNA binding protein, is significantly elevated in the serum in patients with HF, and positively correlates with NT-proBNP and negatively correlates with LVEF [37]. Galectin-3 (Gal-3), a beta-galactoside-binding animal lectin, is a potential novel therapeutic target for inflammatory diseases [38]. The level of Gal-3 is elevated in the plasma in patients with HF and positively correlates with the risk of death and all-cause mortality [39]. Pharmacological inhibition of Gal-3 alleviates cardiac inflammation and fibrosis and ameliorates the HF phenotype in mouse models of HF [40]. Soluble suppression of tumorigenesis-2 (sST2), a biomarker associated with inflammation, has also been reported to be associated with the prognosis of HFrEF [41] and HFpEF [42]. In addition, carbohydrate antigen 125 (CA-125), a high molecular weight glycoprotein, is associated with increased pro-inflammatory cytokines in the serum in patients with chronic HF [43]. Notably, a recent clinical trial has indicated that CA-125 is prognostic for only HFrEF but is not associated with HFpEF [44]. These novel inflammatory markers, which have not been extensively studied in HF, may be potential targets for the inflammation in HF in the future.
Roles of Inflammation in Heart Failure
Elevated local and systemic inflammation is associated with deterioration in cardiac function in both HFrEF and HFpEF. HFmrEF and HFrEF are generally believed to be significantly associated with markers of myocardial traction, whereas HFpEF is more closely associated with biomarkers of inflammation. The clinical features and drug strategies for treating HFmrEF are similar to those for HFrEF [45]. Therefore, we focused on HFrEF and HFpEF (Figure 2).
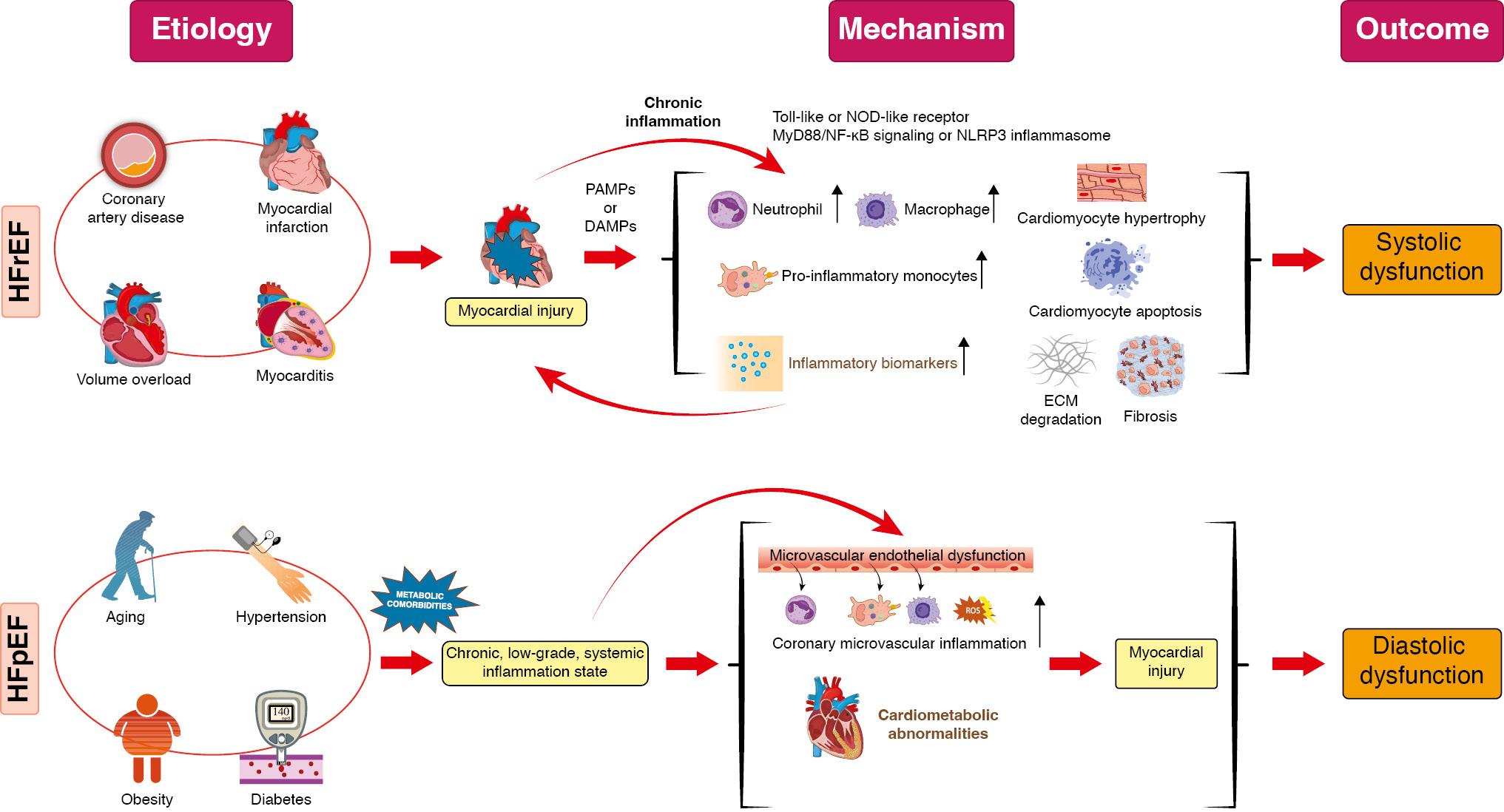
The role of inflammation in HFrEF and HFpEF.
The etiologies of HFrEF and HFpEF are very different. HFrEF is mainly caused by local diseases such as coronary heart disease, hypertension driven stress load, and cardiomyopathy to induce myocardial damage, while HFpEF is more likely to be driven by systemic diseases such as obesity, hypertension, diabetes and aging, which are usually accompanied by systemic chronic inflammation. The myocardial injury in HFrEF appears to precede inflammation, whereas myocardial damages in HFpEF tend to follow inflammation. The pathogen-associated molecular models (PAMPs) and danger-associated molecular patterns (DAMPs) are involved in the inflammatory initiation of HFrEF, activating innate immunity by toll-like or NOD-like receptor. The activated leukocytes can secrete a large number of inflammatory cytokines and chemokines via intracellular myeloid differentiation factor 88 (MyD88), NF-κB signaling pathway or NLRP3 inflammasome, inducing local infiltration and migration, secreting matrix metalloproteinases (MMPs) and degrading myocardial extracellular matrix (ECM). Persistent, excessive inflammation can further damage cardiomyocyte, forming a vicious cycle, and eventually resulting in ventricular systolic dysfunction. The chronic systemic inflammation driven by multiple comorbidities in HFpEF triggers microvascular endothelial dysfunction, leukocyte infiltration, reactive oxygen species (ROS) production, and myocardial metabolic dysfunction, which further cause myocardial damage and resulting in ventricular diastolic dysfunction.
HFrEF
HFrEF is driven largely by inflammation-induced damage and volume overload. Cardiomyocyte damage and loss is the primary driver of HFrEF. The factors initiating a cardiac inflammatory response in HFrEF include pathogen-associated molecular models, such as virus or bacterial antigens, and danger-associated molecular patterns (DAMPs) such as ATP, mtDNA, and hyaluronic acid (released in myocardial ischemia or hemodynamic overload). In fact, most HFrEF-associated inflammation is aseptic, thus indicating activation of innate immunity [46]. Under such sterile conditions, the release of DAMPs triggers an inflammatory response by activating Toll-like receptor 2 (TLR2) or TLR4 on neutrophils and macrophages via intracellular myeloid differentiation factor 88 (MyD88), mitogen-activated protein kinases (MAPK), and NF-κB signaling in local heart tissue [40]. These DAMPS also activate NOD-like receptors on immune cells, such as NLRP3 inflammasomes, thus leading to the release and conversion of pro-IL-1β to IL-1β by caspase-1 [47]. In general, myocardial injury due to multiple causes can trigger a local immune response; lead to leukocyte recruitment and cytokine secretion; elicit cardiomyocyte death, myocardial extracellular matrix degradation, myocardial hypertrophy, or fibrosis; and eventually result in a major loss of contractile function.
HFpEF
The etiology of HFpEF is complex. Obesity, hypertension, diabetes, and aging are common comorbidities with HFpEF [48, 49] that usually precede HFpEF development. The visceral white adipose tissue in obese individuals induces infiltration of monocytes and macrophages, which secrete pro-inflammatory cytokines, adipokines, and immunoglobulins, thus resulting in a low-level chronic systemic inflammatory state [50, 51]. Bariatric surgery decreases systemic inflammation, thereby diminishing the risk of cardiovascular disease [52]. Similarly to obesity, hypertension and aging can also drive systemic microvascular inflammation [53, 54], which participates in the pathogenesis of HFpEF. The primary cardiac changes in HFpEF have been postulated to be coronary microvascular endothelial dysfunction, oxidative stress, and inflammation. Elevated peripheral blood inflammatory biomarkers are more pronounced in HFpEF than in HFrEF, thereby suggesting a more central role of inflammation in HFpEF than in HFrEF [55]. Further study is necessary to elucidate how systemic inflammation transforms into myocardium inflammation in HFpEF, and eventually triggers cardiac diastolic dysfunction.
Clinical Trials Targeting Inflammation Strategies in Heart Failure
Targeted Cytokine Therapies
Pro-inflammatory cytokines play important roles in the progression of chronic HF. Unfortunately, the net results of large-scale clinical trials targeting TNF-α have been disappointing. In 2003, data for the ATTACH trial, the first randomized controlled trial (RCT) of short-term TNF-α antagonism (infliximab), indicated that this treatment did not provide any beneficial effects, and high doses (10 mg/kg) adversely affected clinical condition in patients with moderate-to-severe chronic HFrEF [56]. In 2004, the RENEWAL trial also indicated that TNF-α antagonism (etanercept) did not yield clinically relevant benefits in terms of mortality and hospitalization rates due to chronic HFrEF [57]. The negative results of two large-scale TNF-α antagonism clinical trials were attributed primarily to the complex interactions in inflammatory cytokine networks; therefore, antagonizing individual cytokines alone is unlikely to provide benefit but may cause drug cytotoxicity. Further clinical studies, have evaluated the role of IL-1β inhibition in HF. In 2016, anakinra, an IL-1 receptor antagonist, was found to decrease CRP plasma levels in patients with acute decompensated HFrEF [58]. In 2017, the REDHART trial showed that administration of anakinra for 12 weeks improved exercise capacity patients with HFrEF [59]. In 2019, the CANTOS trial demonstrated that administration of canakinuma, an interleukin-1β inhibitor, decreased HF-associated hospitalizations and mortality among patients with HFrEF, in a dose-dependent manner [60]. The REDHART2 trial is currently assessing the effects of anakinra administration for 24 weeks on cardiorespiratory fitness and HF re-hospitalization in patients with recent hospitalization due to acute decompensated HFrEF [61]. Importantly, the main participants in clinical trials have been patients with HFrEF. As previously stated, chronic systemic inflammation plays a more important role in the development of HFpEF than HFrEF [55]. In 2014, the D-HART trial evaluated the clinical value of anakinra in HFpEF. Administration of anakinra for 14 days was found to improve aerobic exercise capacity in patients with HFpEF [62]. However, administration of anakinra for 12 weeks did not improve ventilation function in patients with HFpEF in the subsequent phase II trial (D-HART2) [63]. In summary, the clinical value of targeting pro-inflammatory cytokines in the treatment of HF must be further studied.
Colchicine
Colchicine, a lipophilic alkaloid extracted from colchicum liliaceae, effectively treats acute gout attacks and has been approved by the US Food and Drug Administration for anti-inflammatory cardiovascular therapy [64]. Multiple clinical trials (COLCOT [65], LoDoCo [66], and LoDoCo2 [67]) have demonstrated the efficacy and safety of low-dose colchicine in patients with acute or chronic coronary artery disease. However, for patients with stable chronic HFrEF, colchicine (0.5 mg bid) decreases inflammatory cytokine levels but has no effect on any clinical indices for HF [68]. Notably, that study was limited by a small sample size. At present, the COLICA trial is evaluating the efficacy and safety of colchicine in the treatment of acute HF, and is expected to provide further clinical evidence [69]. In addition, an animal study has indicated that colchicine alleviates inflammation and ameliorates cardiac diastolic dysfunction in a rat HFpEF model [70]. Currently, the COLpEF trial (NCT04857931), testing the efficacy and safety of colchicine in patients with HFpEF, is ongoing.
Statins
The beneficial effects of statins in cardiovascular diseases have been generally recognized to be independent of their lipid-lowering effects, and to potentially be associated with anti-inflammatory effects [71]. Data on the effects of statins in patients with HFrEF are controversial. In 2006, an RCT reported that atorvastatin decreases serum markers (high sensitivity CRP, IL-6, and TNF-α) of inflammation and ameliorates left ventricular systolic function in patients with HFrEF [72]. However, in 2007, the CORONA trial, the first large-scale, prospective clinical trial evaluating the anti-inflammatory effects of statins in patients with HFrEF, showed that rosuvastatin decreased levels of high-sensitivity CRP and the number of cardiovascular hospitalizations, but did not decrease coronary outcomes or death from cardiovascular causes [73]. Similarly, in 2008, the GISSI-HF trial showed that rosuvastatin did not improve the prognosis of patients with HFrEF [74]. In contrast, a recent meta-analysis has indicated decreased mortality after statin treatment in patients with HFrEF [75]. Current clinical guidelines do not recommend routine use of statins in patients with HFrEF. Many observational studies have consistently reported positive effects of statins on mortality in patients with HFpEF [76–80]. However, statin therapy for HFpEF has not been evaluated in randomized trials. Thus, challenges persist in using statins to treat HF.
SGLT2 Inhibitors
Sodium glucose cotransporter 2 (SGLT2) inhibitors are novel antihyperglycemic drugs demonstrated to be beneficial in cardiovascular disease. Cardiovascular protection by SGLT2 inhibitors has been partly attributed to anti-inflammatory effects [81]. SGLT2 inhibitors indirectly decrease systemic inflammation by modulating inflammatory pathways [81, 82]. SGLT2 inhibitors also exert direct anti-inflammatory effects by modulating immune cells [83]. A meta-analysis of 30 animal studies has suggested that SGLT2 inhibitors decrease inflammatory markers [84]. Two meta-analyses of RCTs have also provided strong evidence supporting the anti-inflammatory effects of SGLT2 inhibitors [85, 86]. The EMPA – REG OUTCOME trial (empagliflozin) [87], CANVAS (canagliflozin) [88], and DECLARE-TIMI 58 (dapagliflozin) [89] trials have consistently shown that SGLT-2 inhibitors significantly decrease severe new-onset HF in patients with type 2 diabetes. Furthermore, the DAPA-HF trial (dapagliflozin) [90] and EMPEROR-Reduced trial (empagliflozin) [91] have provided strong evidence that SGLT2 inhibitors decrease the risk of cardiovascular death and hospitalization among patients with HFrEF, regardless of the presence or absence of diabetes. Two RCTs, the EMPEROR-Preserved trial [92] and DELIVER trial [93], have shown that SGLT2 inhibitors decrease the risk of cardiovascular death and hospitalization among patients with HFmrEF or HFpEF, regardless of the presence or absence of diabetes. Current clinical guidelines strongly recommend the use of SGLT2 inhibitors in patients with HFrEF and HFpEF [94]. Overall, SGLT2 inhibitors have emerged as promising therapeutic agents for targeting inflammation in HF.
Glucagon-Like Peptide 1 Receptor Agonists
Glucagon-like peptide-1 (GLP-1) is released from gut enteroendocrine cells, promotes blood glucose homeostasis, slows gastric emptying, and decreases appetite; its receptor (GLP-1R) agonist has been approved for the treatment of type 2 diabetes and obesity. Accumulating evidence suggests that GLP1-R agonists also exert anti-inflammatory effects [95, 96]. Currently, although three important RCTs, the albiglutide trial [97], FIGHT trial [98], and LIVE trial [99], have shown that GLP1R agonists do not confer beneficial effects in patients with HFrEF, they have been found to decrease CRP concentrations and ameliorate symptoms, physical limitations, and exercise function in patients with HFpEF in STEP-HFpEF trial [100, 101].
Omega-3
Omega-3 fatty acids, found in fish oil, are essential polyunsaturated fatty acids that are considered potential anti-inflammatory agents. Fish oils have been found to decrease TNF-a production in patients with HF [102]. In the GISSI-HF trial [103, 104], a large-scale RCT, omega-3 supplementation significantly decreased all-cause mortality by 9%, and all-cause mortality or cardiovascular hospitalizations by 8%, over 3.9 years in patients with HFrEF. No clinical trials have evaluated the efficacy of omega-3 supplementation for HFpEF, but omega-3 serum levels are inversely associated with cardiometabolic risk factors in patients with HFpEF [105]. Current guidelines recommend omega-3 treatment for patients with NYHA II to IV HF, to decrease the risk of cardiovascular hospitalization and death [94]. Importantly, whether omega-3 fatty acids provide clinical benefit in cardiac disease remains controversial. In 2018, the REDUCE-IT trial [106] showed that omega-3 supplementation significantly decreased cardiovascular risk in patients with elevated triglyceride levels. In contrast, in 2020, the OMEMI trial [107] and STRENGTH trial [108] indicated that high-dose omega-3 did not confer significant cardiovascular risk benefits in patients with myocardial infarction or high cardiovascular risk. Subsequently, a meta-analysis indicated that omega-3 fatty acids decrease cardiovascular mortality and improve cardiovascular outcomes, but increase incident atrial fibrillation [109]. In addition, in the general population, regular use of fish oil supplements might increase risk of atrial fibrillation and stroke [110]. Therefore, further research is needed to evaluate the roles of this treatment in cardiovascular disease.
Vagus Nerve Stimulation
In the pathophysiology of HF, the activity of the vagus nerve is clearly inhibited, thus potentially leading to the release of many inflammatory factors and further aggravating HF. The anti-inflammatory effects of vagus nerve stimulation (VNS) have been well established. Preclinical studies have demonstrated that VNS markedly ameliorates the phenotype of HF through its anti-inflammatory effects in animal models of HFrEF [111, 112] and HFpEF [113]. De Ferrari et al. [114] first reported a clinical trial of VNS for HF, indicating that VNS safely improves quality of life and cardiac function in patients with HFrEF. The safety and efficacy of VNS in patients with HFrEF were further confirmed in the subsequent ANTHEM-HF trial [115]. These beneficial effects have been found to be sustained across multiple years during high-intensity VNS [116]. However, two RCT studies, the NECTAR-HF trial [117] and INOVATE-HF trial [118], have reported that VNS did not improve cardiac function, or decrease the rates of death or worsening of HF, in patients with HFrEF. Clinical studies on VNS in the treatment of HFrEF have yielded controversial findings. Differences in patient demographics, neurologic targets, technology platforms, and VNS mode and delivery might explain the differing results among clinical trials [119]. Recently, a proof-of-concept pilot study has demonstrated that low-level tragus stimulation (LLTS), a noninvasive VNS strategy, attenuates systemic inflammation and provides beneficial effects in patients with acute HF [120]. In addition, LLTS acutely ameliorates left ventricular global longitudinal strain in patients with HFpEF [121]. LLTS for 3 months decreases inflammatory cytokines and improves quality of life, but does not affect echocardiographic markers of diastolic dysfunction in patients with HFpEF [122]. Similarly, in the recent ANTHEM-HFpEF trial, VNS improved clinical symptoms and quality of life, but did not alter mechanical function measures in patients with HFpEF [123]. Importantly, the small sample sizes of those prior clinical studies limited their power to detect differences. Further large-scale randomized clinical trials are required to evaluate the long-term efficacy of VNS in terms of clinical outcomes in patients with HFpEF.
Historical Small-Scale Studied Targets
Methotrexate, a nonspecific anti-inflammatory drug for rheumatoid arthritis, has anti-inflammatory effects and improves NYHA scores, but does not alter LVEF and clinical outcomes in patient with HFrEF [124, 125]. Thalidomide, a drug with potential immunomodulating and matrix-stabilizing properties, ameliorates LVEF in patients with HFrEF [126]. However, positive results of thalidomide in HFrEF have not been found in a subsequent trial [127]. In addition, oxypurinol [128] and allopurinol [129], xanthine oxidase (XO) inhibitors, have not been observed to produce clinical improvements in patients with HFrEF. Despite numerous failed attempts, the search for anti-inflammatory strategies for HF continues. Because of the many available studies, several small clinical studies of anti-inflammation in HF are not discussed herein.
Summary and Future Prospects
Causal evidence has demonstrated that inflammation plays a major role in the development of HF. Targeting inflammation in HF is a topic of an ongoing research. Targeting inflammatory biomarkers in HF remains an important future research direction. Broad-spectrum anti-inflammatory strategies may achieve more significant clinical effects than inhibition of individual pro-inflammatory cytokines. Importantly, most early completed clinical trials assessing inflammation targeting therapies in HF have not yielded promising findings, but recent trials of anti-inflammatory treatment, such as anti-IL-1β therapy, have yielded encouraging preliminary findings. The relevant clinical trials are summarized in Table 1. Developing a specific anti-inflammatory therapy for HF has proven challenging. Well-designed and expensive clinical trials with long follow-up are required for identifying candidate anti-inflammatory therapies with clinical benefits in human HF. Current animal models cannot accurately recapitulate the pathophysiologic heterogeneity in HF, particularly HFpEF, thus challenging the successful translation of conclusions from animal experiments into clinical practice.
Overview of Clinical Trials Targeting Inflammation in Patients with Heart Failure.
| Study (year) | Therapy | Study type | Number of patients | Main HF category | Follow-up | Outcomes |
|---|---|---|---|---|---|---|
| ATTACH (2003) [56] | Infliximab (TNF) | RCT | 150 | HFrEF | 7 months | Did not improve clinical outcomes |
| RENEWAL (2004) [57] | Etanercept (TNF) | RCT | 2048 | HFrEF | 5.7/12.9 months | Did not improve clinical outcomes |
| Van Tassell et al. (2016) [58] | Anakinra (IL-1β) | RCT | 30 | ADHF (HFrEF) | 14 days | Decreased CRP plasma levels |
| REDHART (2017) [59] | Anakinra (IL-1β) | RCT | 60 | HFrEF | 3 months | Increased exercise capacity |
| CANTOS (2019) [60] | Canakinumab (IL-1β) | RCT | 10,061 | HFrEF | 3.7 years | Decreased HF-associated hospitalizations and mortality in a dose-dependent manner |
| D-HART (2014) [62] | Anakinra (IL-1β) | RCT | 12 | HFpEF | 14 days | Decreased systemic inflammatory response; improved aerobic exercise capacity |
| D-HART2 (2018) [63] | Anakinra (IL-1β) | RCT | 31 | HFpEF | 3 months | Decreased plasma CRP levels; did not improve ventilation function |
| Deftereos et al. (2014) [68] | Colchicine | RCT | 267 | HFrEF | 6 months | Decreased inflammatory cytokine levels; did not affect any clinical indices |
| Sola S et al. (2006) [72] | Atorvastatin | RCT | 108 | HFrEF | 12 months | Decreased inflammatory cytokine levels; improved left ventricular systolic function |
| CORONA (2007) [73] | Rosuvastatin | RCT | 5011 | HFrEF | 2.7 years | Decreased high-sensitivity CRP levels and the number of cardiovascular hospitalizations; did not decrease coronary events and cardiovascular death |
| GISSI-HF (2008) [74] | Rosuvastatin | RCT | 4574 | HFrEF | 3.9 years | Did not improve clinical outcomes |
| Fukuta H et al. (2005) [76] | Statins | Observational study | 137 | HFpEF | 21 ± 12 months | Improved survival |
| Roik et al. (2008) [78] | Statins | Observational study | 146 | HFpEF | 1 year | Decreased mortality and rehospitalization rates |
| Tsujimoto and Kajio (2018) [80] | Statins | Observational study | 3378 | HFpEF | 3.3 years | Decreased risk of all-cause and cardiovascular mortality |
| Nochioka et al. (2015) [79] | Statins | Observational study | 3124 | HFpEF | 3.4 years | Improved mortality rates |
| DAPA-HF (2019) [90] | Dapagliflozin (SGLT2 inhibitors) | RCT | 4744 | HFrEF | 18.2 months | Decreased risk of worsening HF or cardiovascular death |
| EMPEROR-Decreased (2020) [91] | Empagliflozin (SGLT2 inhibitors) | RCT | 3730 | HFrEF | 16 months | Decreased risk of cardiovascular death or hospitalization for heart failure |
| EMPEROR-Preserved (2021) [92] | Empagliflozin (SGLT2 inhibitors) | RCT | 5988 | HFpEF | 26.2 months | Decreased risk of cardiovascular death or hospitalization |
| DELIVER (2022) [93] | Dapagliflozin (SGLT2 inhibitors) | RCT | 6263 | HFpEF | 2.3 years | Decreased risk of worsening heart failure or cardiovascular death |
| Lepore et al. (2016) [97] | Albiglutide (GLP-1R agonist) | RCT | 82 | HFrEF | 12 weeks | Did not improve cardiac function |
| FIGHT (2016) [98] | Liraglutide (GLP-1R agonist) | RCT | 300 | HFrEF | 180 days | Did not affect the primary endpoint |
| LIVE (2017) [99] | Liraglutide (GLP-1R agonist) | RCT | 241 | HFrEF | 24 weeks | Did not affect left ventricular systolic function; increased risk of serious cardiac adverse events |
| STEP-HFpEF (2024) [100] | Semaglutide (GLP-1R agonist) | RCT | 529 | HFpEF | 52 weeks | Decreased CRP plasma levels; ameliorated HF-related symptoms |
| GISSI-HF (2008) [103] | Omega-3 | RCT | 6975 | HFrEF | 3.9 years | Decreased all-cause mortality by 9% and all-cause mortality or cardiovascular hospitalization by 8% |
| De Ferrari et al. (2011) [114] | VNS | Open-label study | 32 | HFrEF | 6 months | Improved quality of life and left ventricular function |
| ANTHEM-HF (2014) [115] | VNS | RCT | 60 | HFrEF | 6 months | Indicated encouraging safety and efficacy measures |
| NECTAR-HF (2015) [117] | VNS | RCT | 87 | HFrEF | 6 months | Did not significantly affect primary and secondary endpoints |
| INOVATE-HF (2016) [118] | VNS | RCT | 707 | HFrEF | 16 months | Did not affect rate of death or HF events |
| Dasari et al. (2023) [120] | VNS | RCT | 19 | ADHF (HFrEF) | 4 days | Attenuated systemic inflammation and provided beneficial effects |
| Tran et al. (2019) [121] | VNS | RCT | 24 | HFpEF | 1 hours | Ameliorated left ventricular global longitudinal strain |
| Stavrakis et al. (2022) [122] | VNS | RCT | 52 | HFpEF | 3 months | Decreased inflammatory cytokines and improved quality of life; did not affect echocardiographic markers of diastolic dysfunction |
| ANTHEM-HFpEF (2023) [123] | VNS | RCT | 52 | HFpEF | 12 months | Improved heart failure symptoms; did not affect mechanical function measures |
| Gong et al. (2006) [124] | Methotrexate | RCT | 62 | HFrEF | 4 months | Did not improve LVEF and adverse events; had anti-inflammatory effects; improved NYHA scores and quality of life |
| METIS (2009) [125] | Methotrexate | RCT | 52 | HFrEF | 3 months | Did not improve inflammation and clinical outcome |
| Gullestad et al. (2005) [126] | Thalidomide | RCT | 56 | HFrEF | 3 months | Improved LVEF |
| Orea-Tejeda et al. (2007) [127] | Thalidomide | RCT | 80 | HFrEF | 6 months | Did not improve LVEF |
| OPT-CHF (2008) [128] | Oxypurinol (xanthine oxidase inhibitor) | RCT | 405 | HFrEF | 6 months | Did not achieve clinical improvements in HF |
| EXACT-HF (2015) [129] | Allopurinol (xanthine oxidase inhibitor) | RCT | 253 | HFrEF | 6 months | Did not improve clinical status, exercise capacity, quality of life, or LVEF |
Note: RCT, randomized controlled trial; SGLT2, sodium glucose cotransporter 2; GLP1R, glucagon-like peptide 1 receptor; VNS, vagus nerve stimulation; HF, heart failure; HFrEF, heart failure with decreased ejection fraction; HFpEF, heart failure with preserved ejection fraction; ADHF, acutely decompensated heart failure; TNF, tumor necrosis factor; IL-1β, interleukin-1β; CRP, C-reactive protein; LVEF, left ventricular ejection fraction.
Clinical trials of anti-inflammatory treatment for HF remain ongoing (Table 2). The REDHART2 (NCT03797001) [61] and COLICA (NCT04705987) [69] trials are being conducted to evaluate the clinical efficacy of anakinra and colchicine for acute decompensated HFrEF, respectively. In addition, AZD4831 (NCT04986202), a myeloperoxidase inhibitor; ziltivekimab (NCT05636176), an IL-6 inhibitor; and colchicine (NCT04857931) are currently in clinical trials to assess their therapeutic value against HFpEF/HFmrEF [130, 131]. With refined understanding of the complex inflammatory networks within the heterogeneous syndrome of HF, as well as the cumulative lessons learned from previous clinical trials, the development of more intelligently targeted anti-inflammatory therapeutic strategies might be possible in the foreseeable future.
Ongoing Clinical Trials of Anti-Inflammatory Therapies in Patients with Heart Failure.
| Trial registration number | Therapy | Category of HF | Numbers of patients | Follow-up |
|---|---|---|---|---|
| NCT03797001 | Anakinra (IL-1β receptor antagonist) | ADHF | 102 | 6-month |
| NCT05636176 | Ziltivekimab (IL-6 inhibitor) | HFpEF | 5600 | 4-year |
| NCT04705987 | Colchicine | ADHF | 278 | 2-month |
| NCT04857931 | Colchicine | HFpEF | No message | 6-month |
| NCT04986202 | AZD4831 (myeloperoxidase inhibitor) | HFpEF | 1480 | 1-year |
Note: HF, heart failure; ADHF, acutely decompensated heart failure; HFpEF, heart failure with preserved ejection fraction.