
Tyrosine Kinase Inhibitors
Tyrosine kinases (TKs) catalyze the transfer of γ-phosphate groups from adenosine triphosphate (ATP) to the tyrosine residues of myriad proteins, thereby resulting in protein phosphorylation. As such, TKs participate in many cellular processes, including intracellular and extracellular signaling, energy metabolism, and cell growth and differentiation. Abnormal TK expression can lead to cell proliferation disorders, which are closely associated with proliferation, invasion, apoptosis, and tumor angiogenesis. For instance, the BCR-ABL (Abelson murine leukemia gene) TK is constitutively activated in people with chronic myelogenous leukemia. Accordingly, inhibition of TKs has become a commonly used strategy for cancer treatment.
TK inhibitors (TKIs) have efficacy in treating chronic myelogenous leukemia, breast cancer, renal cell carcinoma, and melanoma [1–3]. Compared with traditional chemotherapeutic drugs, small-molecule TKIs have high efficiency and low toxicity, and therefore have become a major direction in the development of clinical antitumor drugs. Two main drug design strategies have been proposed to inhibit TK-associated oncogenic signaling pathways: (1) antibodies that inhibit activation of the tyrosine kinase receptor, such as trastuzumab directed against HER-2; and (2) small molecule TKIs that cross the cell membrane and target multiple kinases by competing with ATP for the ATP binding site of protein tyrosine kinases. Because these binding sites are highly conserved across kinase families, achieving target specificity is challenging, and off-target effects are common [4, 5]. Although multitargeted kinase inhibition can enhance coordinated effects in the regulation of cancer cell growth, differentiation, and apoptosis, this treatment also poses a substantial risk of adverse effects, owing to the unpredictable mechanisms underlying the anti-cancer effects [6–8].
With the wide application of TKIs in cancer treatment, related cardiovascular adverse reactions have received considerable attention. Clinical data have indicated that arrhythmias, particularly atrial fibrillation (AF), are a common cardiovascular adverse effect of TKIs [9–11] (Table 1). For example, ibrutinib, a first-in-class covalent inhibitor of Bruton’s TK that is commonly used to treat chronic lymphocytic leukemia, has been found to induce new-onset AF in 5% of patients in a phase 3 clinical trial. Multiple subsequent clinical studies have reported an incidence of AF during ibrutinib treatment of 4.8–16% [12], with a relative risk of AF of 3.9 [13].
Arrhythmic Adverse Effects of Various Tyrosine Kinase Inhibitors.
| Medication | Common type (%) | Rare type | Prolonged QTc (>500 ms), % | TdP, % |
|---|---|---|---|---|
| Cobimetinib | AF (3) | NA | 1.2 | 1.2 |
| Dasatinib | SVT (13) | PVC, VT | 0.96 | NA |
| Ibrutinib | AF (4.8–16) | PVC, VT | NA | NA |
| Imatinib | NA | AF, SCD | NA | NA |
| Lapatinib | NA | AF, SCD, SVT | 6.2 | NA |
| Nilotinib | NA | AF, AVB, SCD | 0.68–1.2 | NA |
| Ponatinib | AF (7) | AVB, SB, SVT | NA | NA |
| Regorafenib | NA | AF | NA | NA |
| Sorafenib | AF (5.1) | SB, SVT | NA | NA |
| Sunitinib | NA | AF, SB | 0.52 | <0.1 |
AF, atrial fibrillation; SVT, supraventricular tachycardia; PVC, premature ventricular complexes/contractions; VT, ventricular tachycardia; SCD, sudden cardiac death; AVB, atrioventricular block; SB, sinus bradycardia.
Atrial Fibrillation
AF is the most common sustained cardiac arrhythmia [14, 15]. According to the Global Burden of Disease study in 2010, an estimated 33.5 million people worldwide have AF, including approximately 20 million males. The prevalence of AF is 3% in adults over 20 years of age [16], and the prevalence and incidence gradually increase with age. In addition, AF aggravates the risk of heart failure, coronary heart disease, and stroke, thereby markedly affecting quality of life [17, 18] and placing considerable economic pressure on society.
The pathogenesis of AF is complex and involves myriad mechanisms and pathways. AF is a complex process involving an interplay among structural and electrical atrial remodeling, metabolic remodeling, autonomic imbalance, and genetic factors, thus leading to electrical activity initiation by ectopic foci and reentry, and ultimately inducing AF [19–21].
Metabolic Remodeling
The heart requires a continuous energy supply to sustain muscle contraction and maintain ionic balance. The primary energy source is fatty acids, and glucose, lactate, ketones, and amino acids also contribute to energy production [22]. This energy generation occurs through two major pathways: mitochondrial oxidative phosphorylation and cytosolic glycolysis. The heart’s metabolic flexibility allows it to adapt its energy substrate utilization according to various factors such as workload, nutrient availability, and hormonal state. In this way, the heart can ensure a constant output of ATP under diverse conditions – a crucial aspect in maintaining cardiac function and preventing metabolic disturbances.
In cardiac diseases, such as arrhythmia and heart failure, metabolic remodeling plays a major role. Metabolic remodeling is characterized by mitochondrial dysfunction and a shift in cardiac substrate utilization [23]. Impaired mitochondrial function, as indicated by decreased levels of high-energy phosphates and mitochondrial respiration rates, is observed in failing hearts. Moreover, the tissue content and activity of respiratory chain complexes are altered in animal models of AF and end-stage human cardiac failure [24–27]. Hence, abnormalities in mitochondrial oxidative phosphorylation are relatively more likely to occur during the late stages of cardiac diseases.
Regarding substrate utilization, heterogeneity in fatty acid uptake and utilization has been observed in patients with cardiac diseases. Whereas some studies have reported a decline in the fatty acid oxidation rate and an increase in glucose utilization [28, 29], others have indicated conflicting results [30]. These discrepancies may be attributable to differences in the type and stage of cardiac diseases across studies. Nevertheless, the hypertrophied heart relies primarily on glucose as an energy substrate. This increase in glycolysis, particularly anaerobic glycolysis, may compensate for the decline in fatty acid oxidation in ATP production. However, whether this compensatory mechanism is sufficient to maintain optimal cardiac function remains unclear. Furthermore, metabolic remodeling influences not only energy production, but also electrical and structural remodeling, thus creating a cycle that contributes to the development of various cardiac diseases.
Mitochondrial Dysfunction
TKIs may Trigger Mitochondrial Dysfunction
Ibrutinib
Ibrutinib, a first-generation covalent inhibitor of Bruton’s tyrosine kinase (BTK), is a first-line therapy for B-cell malignancies. Ibrutinib-associated AF toxicity may result from increased reactive oxygen species (ROS) production and modification of CaMKII via oxidation or phosphorylation, thus leading to disrupted calcium release in the sarcoplasmic reticulum (SR), atrial fibrosis, and atrial hypertrophy. Moreover, morphological changes in mitochondria have been observed in ibrutinib-treated mouse cardiomyocytes [31], thereby increasing AF susceptibility, whereas these effects are reversed by apocynin, an NADPH oxidase (NOX) inhibitor.
Sunitinib
Sunitinib is an orally active multitargeted kinase inhibitor with an oxindole structure that selectively inhibits a range of receptor TKs, including vascular endothelial growth factor receptor (VEGFR) type 1/2, platelet-derived growth factor receptors (PDGFR-alpha and PDGFR-beta), stem cell factor receptor (KIT), FMS-like tyrosine kinase-3 (FLT3), glial cell-line-derived neurotrophic factor receptor (RET), and macrophage-colony stimulating factor receptor (CSF1R). Patients receiving sunitinib exhibit symptoms of cardiac systolic dysfunction [32]. Sunitinib also has been found to induce mitochondrial injury and myocardial apoptosis in mouse and rat cardiomyocytes [33]. Mechanistically, sunitinib disrupts cellular energy homeostasis by inhibiting the AMPK pathway, thus causing mitochondrial dysfunction and ultimately chemotherapeutic cardiotoxicity. Mitochondrial dysfunction is characterized by damage to respiratory chain function and diminished ATP production [34].
After 2 weeks of sunitinib treatment, the activity of electron transport chain (ETC) enzyme complexes significantly decreases, and mitochondrial ROS increase. However, co-treatment with mito-TEMPO, a mitochondrial-specific ROS scavenger, for 24 h prevents sunitinib-induced ATP and GSH depletion and increases H2O2 and caspase-3/7 activity in H9c2 cells [35].
Sorafenib
Sorafenib is a multitargeted TKI that inhibits tumor cell proliferation by suppressing the kinase activity of the B-Raf, Raf-1, and Ras/Raf/MEK/ERK signaling pathways. Additionally, it inhibits angiogenesis and induces apoptosis in tumor cells. This drug was granted FDA approval for the management of late-stage renal cell carcinoma in 2006, and in 2007 it became the sole targeted therapeutic option for advanced-stage hepatocellular carcinoma. At clinically relevant concentrations, sorafenib directly impairs the mitochondrial function of H9c2 cardiomyoblasts, as evidenced by the uncoupling of oxidative phosphorylation or inhibition of ETC components [36]. Moreover, ferroptosis, a novel form of cell death distinct from apoptosis and autophagy, is also associated with sorafenib-induced cardiotoxicity and is characterized by mitochondrial morphological changes, such as unusually small mitochondria, elevated mitochondrial membrane density, few or absent mitochondrial cristae, and rupture of the outer mitochondrial membrane [37, 38].
Mechanistically, a compensatory increase in glycolysis occurs after the inhibition of mitochondrial oxidative phosphorylation in sorafenib-treated human-induced pluripotent stem cells. Although this glycolysis ensures a short-term energy supply for cardiomyoblasts, adverse effects may arise in the long term [39]. Moreover, the ROS oxidative stress and cytotoxicity induced by sorafenib treatment is attenuated by c-Met, through activation of the nuclear factor erythroid 2-related factor 2 (Nrf2)-HO-1 antioxidant pathway. Nrf2 is the core regulator of the mitochondrial biogenesis process, whereas c-Met has a protective role by promoting the nuclear translocation of Nrf2 and preventing Nrf2 from binding the inhibitory protein Keap1. Moreover, silencing of Nrf2 weakens the protective function of c-Met against sorafenib-induced mitochondrial toxicity [40].
Imatinib
Imatinib, a first-generation BCR-ABL TKI, also exhibits affinity toward PDGFRs and the c-KIT gene. However, mitochondrial toxicity in cardiomyocytes has been reported after imatinib treatment in vivo and in vitro. For example, Kerkelä et al. have reported NYHA class 3–4 heart failure progression in patients receiving imatinib therapy (7.2 ± 5.4 months). Ultrastructural analysis has further revealed pleomorphic mitochondria with effaced cristae, as often observed in hearts with impaired mitochondrial function. Moreover, in vitro, imatinib induces the collapse of the mitochondrial membrane potential in isolated cardiomyocytes in a dose-dependent manner, partly because of PERK-eIF2α activation and the accumulation of downstream unfolded proteins in the mitochondrial intermembrane space, thus ultimately inducing mitochondrial death pathways [41, 42].
Mitochondrial toxicity of imatinib has also been observed in C2C12 murine myoblasts and myotubes, and in human rhabdomyosarcoma cells [43, 44]. Imatinib exhibits cytotoxicity toward C2C12 cells with a half maximal inhibitory concentration of 20 μM, by inhibiting OXPHOS complexes and core transporters of glucose/nucleoside uptake, and producing superoxides. This imatinib-associated alteration in energy metabolism and disrupted mitochondrial function may partly account for TKI-induced fatigue.
Mitochondrial Dysfunction and AF
Understanding the Intricacies of Mitochondrial Physiology in Cardiomyocytes
Mitochondria occupy approximately 30% of the cardiomyocyte volume. Hence, the biological functions of cardiomyocytes are closely associated with mitochondrial activity. The heart is a highly energy-consuming organ, and its proper pump function requires mitochondrial aerobic oxidation to produce sufficient energy. Free fatty acids and glucose, the main energy-supplying substances, are converted to acetyl-CoA via fatty acid β-oxidation and aerobic oxidation of glucose [45]. Subsequently, acetyl-CoA enters the tricarboxylic acid cycle within the mitochondria, and NADH and FADH2 are generated. Each NADH generates three ATPs, whereas each FADH2 generates two ATPs through the respiratory chain. During the transfer of electrons from NADH/FADH2 to oxygen, electron leakage in respiratory chain complexes I or III allows oxygen to directly accept single electrons and generate superoxide anion radicals, which can further generate ROS, such as hydroxyl radicals and hydrogen peroxide [46, 47].
Mitochondria can acutely monitor changes in intracellular calcium concentration, which is dynamically regulated via influx process mediated by mitochondrial Ca2+ uniporter (MCU) and efflux process mediated by sodium/calcium exchange (NCX), the Ca2+ antiporter, and mitochondrial permeability transition pore (MPTP) [48, 49], thereby maintaining a relatively constant intracellular concentration of calcium. Mitochondria are highly sensitive to changes in nutrient and oxygen requirements; depending on the environment, these changes can be compensated for until decompensation occurs. Mitochondrial dysfunction is an important pathogenic factor in various cardiovascular diseases and is closely associated with AF [25, 50–52].
Diminished ATP Production
In addition to supporting contractile activity, one-third of the ATP produced in cardiomyocytes is used to maintain the function of ion channels and transporters on the cell membrane and SR. The sarcolemmal ATP-sensitive K+ (sarcK ATP) channel is highly sensitive to intracellular energy metabolism, and its extent of channel activation depends on the mitochondrial transmembrane potential (ΔΨm) [53]. When mitochondrial function is impaired, the decrease in ATP production fails to maintain ΔΨm, thus decreasing mitochondrial transmembrane potential and increasing the frequency of sarcK ATP channel opening. Consequently, the conduction of myocardial regional electrical activity may slow, and overall electrical heterogeneity may increase, thereby providing conditions for the development of reentry [54].
In addition, diminished mitochondrial ATP synthesis weakens the physiological function of the Na+/K+ pump and the ATP-dependent Ca2+ pump, thus resulting in intracellular calcium overload and promoting AF [55].
Overproduction of ROS
Mitochondrial dysfunction can lead to impaired ETC function. Increased electron leakage results in ROS overproduction that cannot be effectively countered by ROS scavenging systems, such as antioxidant enzymes and nonenzymatic factors [47, 56]. Overproduction of ROS impairs mitochondrial respiratory chain complex enzyme activity, thus decreasing ATP generation capacity. In addition, ROS act on cardiac systolic and diastolic function-related proteins, and consequently impairs electrical, mechanical, biochemical, and metabolic functions in cardiomyocytes. Moreover, through MPTP overactivation, excessive ROS can trigger amplified ROS production after oxidative stress, in a process termed ROS-induced ROS release [57].
The link between ROS overproduction and AF has been confirmed experimentally. In atrial tissue from patients with AF, Bukowska et al. have reported mitochondrial structure and respiration impairment and oxidative stress activation, accompanied by an increase in oxidative stress-related markers (HO-1 and LOX-1) These mitochondrial toxicity phenotypes have been recapitulated and validated in 24-hour rapidly paced human atrial tissue slices [58]. Xie et al. have also found markedly greater oxidative stress and ROS production in the atrial myocytes of patients with chronic AF than in those with sinus rhythm, thus increasing oxidation of type 2 ryanodine receptor (RyR2). RyR2 is a calcium channel that governs the release of Ca2+ from the SR, whose oxidation contributes to increased Ca2+ leakage. Pathological Ca2+ release from the SR triggers inward Na+/Ca2+ exchange currents, delayed afterdepolarizations (DADs), and ultimately atrial electric remodeling [59]. ROS also activate NF-κB and its downstream signaling pathways through the mitogen-activated protein kinase pathway [60], stimulate inflammatory factor production, and subsequently cardiomyocyte fibrosis and structural remodeling, thereby serving as the pathophysiological basis of AF.
Dysregulation of Calcium Homeostasis
Mitochondrial dysfunction disrupts calcium regulatory function, thus causing intracellular calcium overload [61], which in turn triggers the irreversible opening of MPTP. MPTP opening enables intracellular component entry to the mitochondria. Disruption of the immediate intra-mitochondrial environment leads to swelling membrane destruction and consequently further aggravates mitochondrial dysfunction. Additionally, MPTP opening causes leakage of intra-mitochondrial contents, including cytochrome c and apoptosis-inducing factor, into the cytosol, thereby activating the downstream apoptotic cascade and leading to myocardial apoptosis [62].
Montaigne reported that patients with postoperative AF have atrial myocardium mitochondria with lower Ca2+ uptake and storage capacity, compared with patients who maintain sinus rhythm after coronary artery bypass grafting [63]. Moreover, Bukowska et al. have found that Ca2+ influx via L-type calcium channels leads to the overexpression of oxidative stress markers and adhesion molecules in patients with cardiac tachyarrhythmia. In ex vivo atrial tissue samples from patients with AF, Ca2+ channel blocker treatment decreases morphological abnormalities, such as mitochondrial swelling, and significantly improves mitochondrial respiratory function [58].
Dysregulation of Mitochondrial Biogenesis
PGC-1⍺ is a crucial regulator of mitochondrial biogenesis. In a goat model of AF induced by rapid atrial pacing, downregulation of the PGC-1⍺/NRF-1/Tfam pathway has been found to promote atrial metabolic remodeling, thus impairing mitochondrial function [64]. Moreover, fenofibrate prevents myocardial metabolic remodeling by regulating the peroxisome proliferator-activated receptor (PPAR)-⍺/sirtuin1/peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1⍺ pathway [65]. A significant decrease in PGC-1⍺ also occurs in the atrial tissue of patients with postoperative AF [66].
mtDNA Mutations
Mitochondria have their own genetic material, double-stranded circular mitochondrial DNA (mtDNA), whose mutation has been found to decrease mitochondrial ATP synthesis and ΔΨm in mice [67]. Moreover, mtDNA mutations have been found to occur in both mtDNA control and coding regions in patients with chronic AF, and are associated with atrial structural and electrical remodeling [68]. Zhang et al. have found that mtDNA copy number is an independent risk factor for postoperative AF, with good sensitivity and specificity [69].
Shift in Cardiac Substrate Utilization
To meet the high energy demands of the beating heart, contracting cardiac myocytes depend primarily on mitochondrial oxidative phosphorylation; however, glycolysis is also an important energy production pathway. Approximately 60–90% of the generated ATP is derived from fatty acid oxidation, whereas the remaining 10–40% is provided by glucose oxidation [70]. AF manifests as rapid, disorganized atrial contraction that increases energy demand [71], and the cardiac substrate utilization pattern changes accordingly [72].
Shift in Cardiac Substrate Utilization caused by TKIs
Sorafenib
Non-targeted metabolite profiling has indicated that sorafenib cardiotoxicity is associated with elevated lactate levels, thus indicating enhanced anaerobic glycolysis in CMs. Moreover, combined losartan and sorafenib therapy reverses the metabolic phenotype, as evidenced by a significant decrease in glycine, urea, and fatty acid levels [73].
Sunitinib
Metabolomic analysis of heart tissue from sunitinib-treated mice has shown downregulation of docosahexaenoic acid, arachidonic acid /eicosapentaenoic acid, O-phosphocolamine, and 6-hydroxynicotinicacid levels. However, cardiotoxicity may also be associated with the depletion of polyunsaturated fatty acids [74]. Moreover, Sayed-Ahmed et al. have reported that sunitinib decreases AMPK and CPT1 expression, potentially via inhibition of mitochondrial functions, including long-chain fatty acid transport and energy generation [75].
The hepatotoxicity of sunitinib is also associated with altered metabolic pathways, including fatty acid β-oxidation, amino acids, nucleotides, lipids, bile acids, and tricarboxylic acid cycle intermediates, after sunitinib treatment [76].
Ibrutinib
Compared with cells poorly responsive to ibrutinib, ibrutinib-sensitive cells show significant improvements in metabolic pathways, such as glycolysis, the pentose phosphate pathway, the TCA cycle, and glutaminolysis, during ibrutinib treatment [77].
Regorafenib
The combination of palbociclib and regorafenib impairs glucose uptake and utilization capacity, and is accompanied by decreased expression of crucial enzymes associated with the glycolytic pathway, such as HK2, PFKP, aldolase A, and PKM2 [78].
Shift in Cardiac Substrate Utilization and AF
Glucose Metabolism
Glucose metabolism accounts for approximately 30% of the total energy production in cardiomyocytes, whereas glucose transporters primarily regulate glucose uptake in cells [79, 80]. Although AMPK/CaMKII activation during AF upregulates GLU-4 expression, the overall metabolic phenotype demonstrates suppression of glucose membrane transport and diminished glucose uptake, owing to the downregulation of SNAP-23 expression [81].
Elevated levels of rate-limiting enzymes associated with the glycolytic pathway have been observed in patients with permanent AF. Similarly, proteomic analysis of atrial tissue from an ovine AF model has revealed upregulation of glycolytic pathway intermediate metabolites (2-phosphoglyceric acid, 1,3-bisphosphoglyceric acid, and pyruvate) and downregulation of aerobic oxidative pathways. Interestingly, this phenomenon is similar to the Warburg effect observed in tumor cells. Moreover, metformin has been found to inhibit the Warburg effect and to ameliorate myocardial fibrosis in a canine AF model, by suppressing the expression of key Warburg effect factors (HIF-1α, PDK1, HK, and LDH), increasing the expression of PDH, decreasing atrial lactate production, and ameliorating metabolic dysregulation in atrial cells [82].
Lipid Metabolism
Fatty acids enter cardiomyocytes via FAT/CD36, whereas carnitine palmitoyl transferase-1 (CPT-1) serves as a transport enzyme facilitating fatty acid entry into mitochondria. Indeed, the fatty acid oxidation pathway is the primary energy source in cardiomyocytes; myocardial hypertrophy, ventricular remodeling, and chronic heart failure are associated with decreased myocardial fatty acid supply [83]. Additionally, children with inherited fatty acid oxidation defects are prone to various arrhythmias, including AF [84].
PPAR and its activator PGC-1⍺ are key regulators of cardiac fatty acid metabolism. The PPAR pathway is associated with myriad diseases, including myocardial fibrosis, diabetes, atherosclerosis, heart failure, and AF [85]. Liu et al. have reported that metformin upregulates the expression of FAT/CD36, CPT-1, and VLCSD in a canine AF model by activating the AMPK/PGC-1⍺/PPAR⍺ pathway, thereby decreasing lipid accumulation in atrial myocytes [65]. Similarly, fenofibrate improves the atrial effective refractory period and decreases the risk of AF by activating the PPAR⍺/sirtuin1/PGC-1⍺ pathway, thus decreasing atrial lipid accumulation [65]. Use of thiazolidinedione PPAR-γ agonists also decreases the incidence of AF in patients with diabetes [86].
Fatty acid-binding protein-3 (FABP-3) is a lipid chaperone that transports fatty acids into the mitochondria. Shingu et al. [87] have observed that new-onset AF during the periprocedural period of cardiac surgery is associated with decreased atrial FABP3 expression. Moreover, Rader et al. have reported that an increase in heart-type fatty acid-binding protein (HT-FABP) abundance is associated with AF after cardiac surgery [88]; these findings suggest that FABP may be a potential therapeutic target for AF.
Metabolic Remodeling in Other TKI-associated Cardiotoxicity and Association with Electrophysiological Toxicity
Traditional chemotherapy approaches in cancer treatment have the potential to cause irreversible harm to cardiomyocytes, thus leading to subsequent acute or chronic left ventricular dysfunction. TKIs, which are commonly used as initial chemotherapy agents for hematologic malignancies and solid tumors, have lower toxicity than conventional chemotherapy drugs. However, they may still induce cardiotoxicity, characterized primarily by diminished cardiac contractility and the development of heart failure [6, 89].
Rennert et al., in a nested case-control analysis, have found that trastuzumab, cetuximab, panitumumab, and sunitinib significantly increase the risk of new-onset heart failure [90]. The cardiotoxicity of TKIs is mechanistically associated with impaired mitochondrial function in cardiomyocytes, perturbed calcium homeostasis, and ROS overproduction, all of which are associated with metabolic remodeling. Furthermore, off-target effects may partially explain TKI-associated heart failure. As observed in the aforementioned study, all EGFR signaling inhibitors increase the risk of heart failure, because EGFR signaling not only plays a crucial role in tumor growth, but also regulates the growth, repair, and survival of cardiomyocytes. Sunitinib uniquely destroys pericytes, which are essential for blood vessel formation and maintenance. This drug might also cause heart failure through off-target effects by directly damaging cardiomyocyte mitochondria, inhibiting the compensatory upregulation of AMPK, and inducing cytochrome C-mediated apoptosis.
In consideration of the off-target effects of TKIs and the mechanisms of cardiac function regulation, brain-derived neurotrophic factor (BDNF) is a key protein that warrants attention. Lafuente et al. have described the critical role of the BDNF-TrkB pathway in ameliorating TKI-mediated drug injury through environmental enrichment [91]. Feng et al. have found that the BDNF/TrkB signaling axis is closely associated with heart development, because cardiac-specific TrkB knockout mice exhibit impaired cardiac contractility. Additional research has revealed that BDNF in normal cardiomyocytes enhances calcium cycling and contractile function through CaMKII, whereas TrkB-deficient cardiomyocytes in heart failure models lack tyrosine kinase activity and are insensitive to BDNF [92]. Recent work by Yang et al. has further demonstrated symptoms of heart failure in cardiac-specific TrkB knockout mice. These mice show diminished exercise capacity and impaired mitochondrial biogenesis with downregulation of PGC-1α, which mediates mitochondrial dysfunction [93]. In summary, BDNF plays a crucial role in normal electromechanical activity and myocardial metabolism, and may serve as a potential therapeutic target for the prevention and treatment of TKI-associated cardiac toxicity.
The relationship between TKI-induced AF and other cardiovascular complications, such as systolic hypertension and left ventricular dysfunction, is an important consideration. Although TKIs have been associated with both AF and cardiovascular complications, whether AF is a consequence of TKI-induced hypertension/systolic dysfunction or is coincidental is currently unclear. Some studies have suggested a potential causal link between TKI-induced AF and hypertension/systolic dysfunction. A single-center retrospective study including 562 patients with B cell malignancies treated with ibrutinib has reported an incidence of new-onset or worsening hypertension of 78.3% during a median follow-up of 30 months. Through multivariate analysis, new-onset or worsening hypertension has been associated with a higher occurrence of other major adverse cardiovascular events (particularly AF) [94]. From a mechanistic standpoint, TKIs can disrupt signaling pathways involved in cardiac function and vascular regulation, thus leading to left ventricular dysfunction and hypertension. These changes may contribute to the development of AF. Furthermore, shared underlying mechanisms, such as oxidative stress and inflammation, may have roles in both AF and the cardiovascular complications induced by TKIs [31].
However, other studies have proposed that TKI-induced AF might be an independent adverse effect unrelated to hypertension/systolic dysfunction. Of note, in patients with concomitant atrial fibrillation, the administration of tyrosine kinase inhibitors can block potassium ion currents, similarly to the effects of quinidine or procainamide, thus shortening atrioventricular node refractoriness [95]. Many patients with cancer receiving tyrosine kinase inhibitors may also experience alterations in their blood electrolyte levels, such as changes in potassium ion concentration. These studies suggest that TKIs directly affect cardiac ion channels and electrical conduction, and consequently lead to atrial electrical remodeling and AF.
More research is needed to fully elucidate the relationship between TKI-induced AF and hypertension/systolic dysfunction. Future studies should investigate the temporal sequence of these complications and explore common underlying mechanisms. Better understanding of these relationships would inform clinical management strategies and guide the development of preventive measures for TKI-induced cardiovascular complications, including AF.
Management of TKI-Associated Arrhythmias
TKI-induced arrhythmia is easily monitored and can serve as an early warning sign for other related consequential cardiovascular disorders. Of note, TKI-associated QT prolongation, particularly that observed with sunitinib, pazopanib, and vandetanib, cannot be neglected. Interestingly, QT interval prolongation is a principal pharmacological mechanism of class III antiarrhythmic drugs, including amiodarone. Effectively balancing the use of anti-atrial fibrillation medications and the risk of QT interval prolongation is particularly important in the clinical management of TKI-associated arrhythmias.
Healthcare providers must carefully assess the risks and benefits of TKI therapy, including the potential for arrhythmias, when considering these medications for their patients. In patients diagnosed with underlying diseases, such as hypertension, coronary heart disease, or diabetes, cardiac function should be closely monitored. Baseline ECGs should be obtained to evaluate the risk of arrhythmia. Furthermore, biochemical markers such as myoglobin (MYO), B-type natriuretic peptide (BNP), and other relevant indexes should be obtained and appropriately managed [9, 96]. Electrolyte levels, particularly sodium, potassium, and calcium, should be monitored and corrected if abnormal [97]. Drug-drug interactions should also be considered. Given that most TKIs undergo metabolism via CYP enzymes and transport via P-gp, the coadministration of other drugs that may influence CYP enzymes or competitively bind P-gp requires careful evaluation. Additionally, drugs known to prolong the QT interval or induce AF should be avoided [98, 99]. Dosing adjustments or discontinuation of TKI treatment is crucial, particularly when the QT interval exceeds 500 ms or changes exceed 60 ms with respect to baseline. Finally, if patients experience symptoms such as fainting, headaches, or irregular heartbeat after medication ingestion, immediate healthcare assistance should be sought. Decisions regarding heart rate or rhythm control should prioritize patient-centric and symptom-oriented approaches. Beta-blockers may be a preferred option for heart rate control, whereas type Ic and type III antiarrhythmic drugs can be beneficial for maintaining heart rhythm stability [6, 100].
Conclusion
TKIs are a new class of potential therapeutic agents for cancer treatment. Clinical and basic research data suggest that TKI treatment may increase the risk of AF, but the mechanisms underlying TKI cardiotoxicity remain elusive. On the basis of current understanding of AF pathogenesis and the pharmacological properties of TKIs, these drugs are believed to induce AF by interfering with myocardial energy metabolism via inducing mitochondrial dysfunction and a shift in cardiac substrate utilization (Figure 1). Understanding the potential mechanisms underlying AF occurrence after TKI treatment will provide important insights for alleviating the arrhythmogenic toxicity of these promising antitumor agents. With the rapid advancement of oncocardiology, collaboration between cardiologists and oncologists is expected to improve the management of patients with cancer.
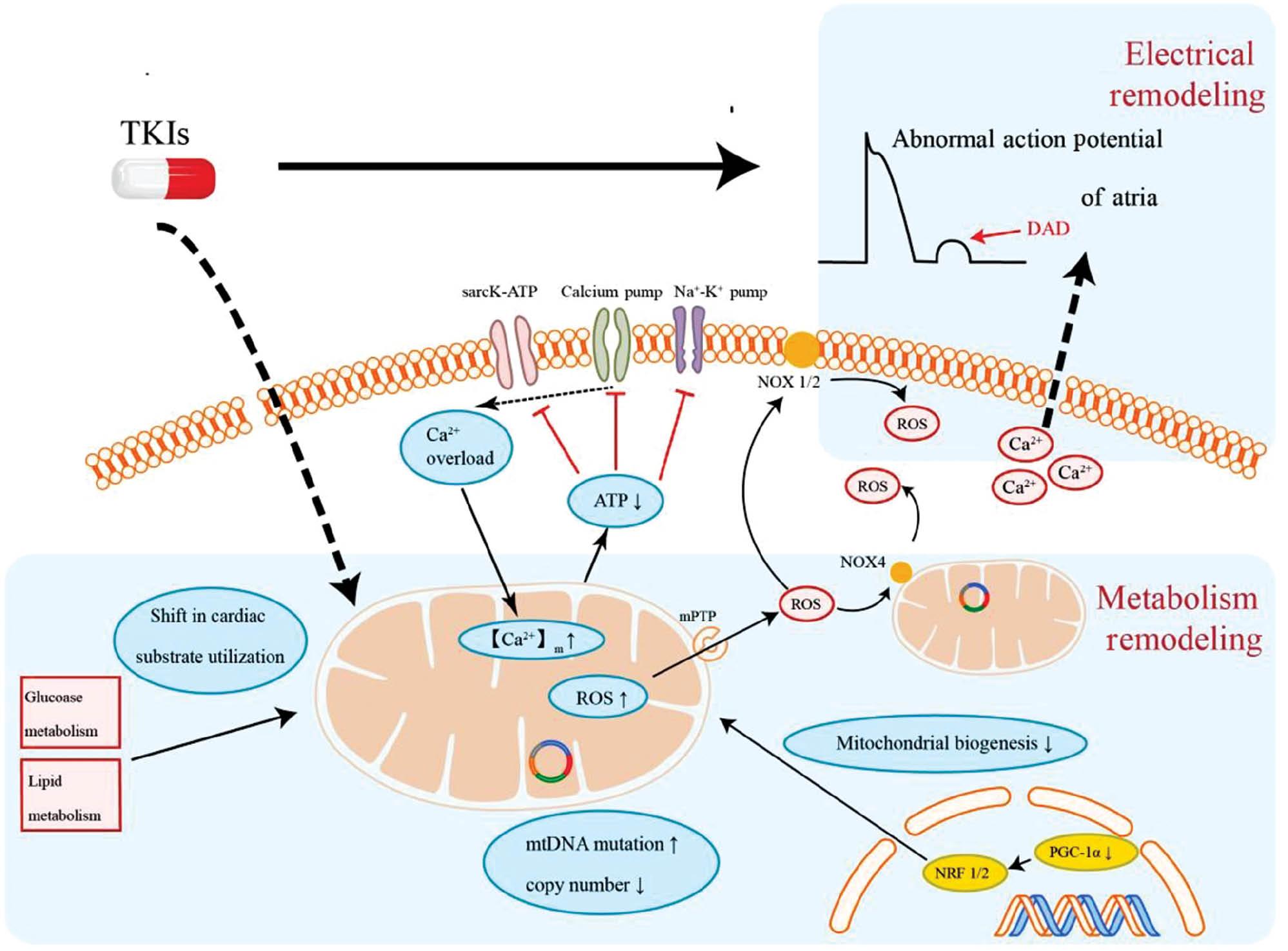
Mechanisms of Metabolic Remodeling in TKI-Induced Atrial Fibrillation.
TKIs exert toxic effects on mitochondria and decrease ATP production. Insufficient energy supply impairs the function of energy-consuming ion pumps and leads to cytosolic calcium overload. As a cellular calcium reservoir, mitochondria regulate cytosolic Ca2+ levels. Upregulated intracellular Ca2+ increases mitochondrial Ca2+ and ultimately mitochondrial Ca2+ overload, thus further exacerbating mitochondrial dysfunction and ROS production. Opening of the mitochondrial permeability transition pore (MPTP) is triggered by mitochondrial Ca2 overload. The subsequent release of ROS via mPTP triggers a transient increase in mitochondrial ROS production, termed ROS-induced ROS release (RIRR). Decreased expression of PGC-1α inhibits the mitochondrial biogenesis process. TKIs also increase the abundance of mtDNA mutations and alter the cardiac substrate utilization pattern. Intracellular Ca2 overload induces DADs. After DADs reach the threshold voltage level, ectopic activity can initiate sustained tachyarrhythmia. The atrial metabolic remodeling process and the subsequent electrical remodeling process are the main causes of TKI-induced atrial fibrillation. TKI, tyrosine kinase inhibitors; ROS, reactive oxygen species; ATP, adenosine triphosphate; mtDNA, mitochondrial DNA; NOX4, NADPH oxidase 4; NRF 1/2, nuclear factor erythroid 2-related factor 1/2; PGC-1⍺, peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1 alpha.