
Significance Statement
The role of N6-methyladenosine (m6A)-associated SNPs in AF remains unknown. Through integrative analysis of a GWAS dataset and m6A-SNPs, we successfully identified 1429 unique m6A-SNPs significantly associated with AF, 17 of which reached genome-wide significance. Gene expression analysis indicated that genes affected by these m6A-SNPs were differentially expressed between AF samples and normal control samples. The results suggest that m6A-SNPs may play an important role in the genetics of AF.
Introduction
Atrial fibrillation (AF) is a prevalent type of cardiac arrhythmia with unclear molecular genetics [1, 2]. Linkage analysis in patients with familial or early-onset AF has identified rare pathogenic variants in ion channel genes such as KCNQ1 and SCN5A [2]. Furthermore, population-based genome-wide association studies (GWAS) have identified numerous rare or common single-nucleotide polymorphisms (SNPs) in more than 100 loci. These findings suggest that, in addition to known common factors, genetic components are involved in the development of AF, and the large amount of available genetic data can accelerate functional research on AF pathogenesis. The identification of genetic variations associated with AF susceptibility can provide a better understanding of underlying pathophysiological mechanisms, aid in the development of more robust risk prediction models and improve therapies. Although most AF-associated SNPs are found in noncoding regions, some of these SNPs have been demonstrated to increase AF risk by regulating the expression levels of effector genes [3–5]. However, most SNPs still require functional characterization.
N6-methyladenosine (m6A) RNA methylation, a prevalent modification in various RNA types, plays essential roles in multiple biological processes, such as RNA-nuclear export, splicing, stability, translation efficiency, RNA-protein interaction and microRNA biogenesis [6]. Recently, research has demonstrated that m6A modification of mRNA is crucial for maintaining cardiac function, and its aberrant regulation is associated with cardiac remodeling, ventricular arrhythmia, ischemia-reperfusion injury and heart failure [7–9]. In addition, a recent study has conducted a systematic evaluation of the RNA modification patterns mediated by differential m6A regulators in AF versus non-AF samples, and six key differential m6A regulators have been identified. These findings have suggested that m6A RNA methylation has a diagnostic value as high as 82.5% in distinguishing patients with AF from healthy participants [10]. Similarly, another study has reported that m6A RNA modification-associated key regulatory genes, including METTL3, RBM15B and METTL14, show significantly higher expression in the left atrial tissues of patients with AF than patients with sinus rhythm, on the basis of data mining analyses [11]. These results suggest that m6A RNA methylation might play a role in the pathogenesis of AF and might be a potential therapeutic target for AF. Further investigation of m6A RNA methylation modification in AF is necessary to uncover its specific roles and mechanisms in the pathogenesis of AF.
Substantial progress in the field of bioinformatics, and the increasing availability of public databases and powerful algorithms, has enabled identification of genomic m6A-related single nucleotide polymorphisms (m6A-SNPs) and their association with various human diseases, such as cancer, cardiovascular diseases, kidney diseases, neurological diseases and immune-associated diseases [12]. Nonetheless, the relationship between m6A-SNPs and AF remains poorly understood. To address this knowledge gap, we conducted an integrative analysis of a published GWAS dataset and a comprehensive list of m6A-SNPs. Our study was aimed at exploring the association between m6A-SNPs and AF, to advance understanding of the molecular basis of AF and identify novel therapeutic targets for this disease.
Methods
Identification of m6A-SNPs in Patients with AF
To identify potential AF-associated m6A SNPs, we acquired two datasets. The first (AF2018, http://csg.sph.umich.edu/willer/public/afib2018/) was a published GWAS dataset composed of 60,620 AF cases and 970,216 controls, which included association P-values between 34,740,186 genetic variants with a minor allele frequency>2.5×10−5 and AF [13]. The second was a list of m6A-SNPs from the m6AVar database (http://m6avar.renlab.org/) [14].
Subsequently, we selected AF associated m6A-SNPs by combining the GWAS summary dataset and the list of m6A-SNPs (Figure 1). Only SNPs with P-values<0.05 were considered in the following analyses. To determine whether the number of m6A-SNPs associated with AF was significantly enriched, we randomly selected non-m6A-SNPs from the AF GWAS dataset 1000 times, then compared the number of non-m6A-SNPs with a GWAS P-value<0.05 in the 1000 randomizations with the number of m6A-SNPs with a GWAS P-value<0.05.

An in silico replication study was conducted with data from a multi-ethnic GWAS on AF that was published in 2018 [15] and is available at http://www.kp4cd.org/datasets/mi. m6A-SNPs were replicated if they were associated with AF with P<0.05 in both GWAS datasets used in this work.
Expression Quantitative Trait Locus Analysis of AF-associated m6A-SNPs
The main function of m6A has been proposed to be the post-transcriptional fine-tuning of gene expression [16]; therefore, m6A may be involved in the progression of AF by regulating gene expression. To determine which m6A-SNPs might result in gene expression changes, we performed expression quantitative trait locus (eQTL) analysis in different cells and tissues by using HaploReg [17], a web-based variant annotation tool available at https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php. eQTLs are empirically divided into two classes: cis and trans. A cis-eQTL coincides with the location of the underlying gene, whereas the observed location of a trans-eQTL does not coincide with the location of the gene.
Differential Expression Analysis
For m6A-SNPs showing eQTL signals, we further investigated whether the expression levels of the genes on which these m6A-SNPs showed eQTL effects were associated with AF. To this end, we first downloaded three normalized expression profile datasets (GSE14975, GSE108660 and GSE2240) from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) [18]. GSE14975 included five atrial tissue samples from patients with AF and five samples from normal controls [19]. GSE108660 detected the expression profiles of mRNAs in five patients with AF and five patients without AF. The GSE2240 dataset contained ten samples obtained from patients with permanent AF and 20 samples from patients in sinus rhythm [20, 21]. Subsequently, for each gene m6A-SNP showed eQTL effects, we performed differential expression analyses between AF and non-AF samples using t tests in R language (Version 3.6.1). Genes with P-values<0.05 were identified as differentially expressed.
Results
Identification of 17 m6A-SNPs Associated with AF with Genome-wide Significance
By mapping 13,703 high, 54,621 medium and 247,269 low confidence level m6A-SNPs in human samples from the m6AVar database to 34,740,186 genetic variants associated with AF in the AF GWAS dataset, we obtained a total of 22,982 unique m6A-SNPs associated with 24,581 m6A sites for AF. For the 22,982 unique m6A-SNPs identified in the GWAS data, 774, 3883 and 18,325 m6A-SNPs were annotated with high, medium and high confidence levels, respectively. Among these m6A-SNPs, 1429 (6.2%) unique SNPs were nominally (P-value<0.05) associated with AF. Seventeen m6A-SNPs had a genome-wide significant (P-value<5×10−8) association with AF (Figure 2). Detailed information for the 17 m6A-SNPs is provided in Table 1. The locations of exonic m6A-SNPs in the target transcripts are shown in Figure 3. For the 17 genome-wide significant AF-associated m6A-SNPs, rs1061259 (P-value=5.48×10−9) and rs1881193 (P-value=2.34×10−8) were mapped with a medium confidence level, and the remaining 15 were mapped with a low confidence level.
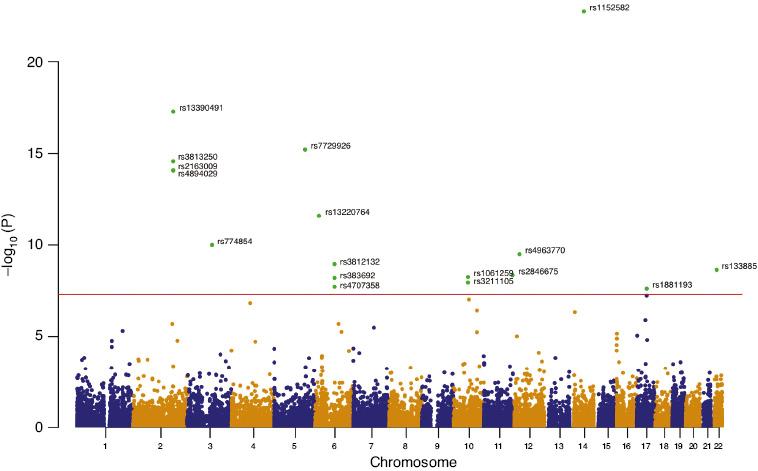
Manhattan Plot of Genome-wide m6A-SNPs Identified in AF Samples (P-value<5×10−8).
The Manhattan plot presents -log10 (P-value) for each of the 22,982 m6A-SNPs associated with AF. SNPs with associated P-values less than 5.0×10−8 are labeled with identifiers.
m6A-SNPs with Genome-wide Significant Associations with AF.
| SNP | Chromosome | Position | Mutation type | P-value | Gene(s) | m6A ID | m6A position | m6A function | Cis-eQTL | Trans-eQTL |
|---|---|---|---|---|---|---|---|---|---|---|
| rs1061259 | chr10 | 65225244 | Exon | 5.48E-09 | JMJD1C-AS1 | m6A_ID_25666 | 65225245 | Loss | 1 | 10 |
| rs1152582 | chr14 | 64692630 | 3′-UTR | 1.75E-23 | SYNE2 | m6A_ID_276113 | 64692631 | Loss | 1 | 9 |
| rs13220764 | chr6 | 18366699 | Exon | 2.40E-12 | piR-50760, piR-35183, piR-50759 | m6A_ID_189225 | 18366697 | Gain | – | – |
| rs133885 | chr22 | 26159289 | Nonsynonymous | 2.22E-09 | MYO18B | m6A_ID_347214 | 26159291 | Loss | – | – |
| rs13390491 | chr2 | 179582327 | Nonsynonymous | 5.00E-18 | TTN | m6A_ID_148344 | 179582345 | Loss | 0 | 2 |
| rs1881193 | chr17 | 44248769 | Synonymous | 2.34E-08 | KANSL1 | m6A_ID_46559 | 44248752 | Loss | 1 | 3 |
| rs2163009 | chr2 | 179455207 | Synonymous | 7.74E-15 | TTN | m6A_ID_147926 | 179455191 | Loss | 0 | 4 |
| rs2846675 | chr11 | 128787036 | 3′-UTR | 4.38E-09 | KCNJ5 | m6A_ID_255272 | 128787035 | Loss | – | – |
| rs3211105 | chr10 | 64945364 | Synonymous | 1.10E-08 | JMJD1C | m6A_ID_235300 | 64945365 | Loss | 1 | 6 |
| rs3812132 | chr6 | 87970301 | Synonymous | 1.08E-09 | ZNF292 | m6A_ID_195511 | 87970328 | Loss | 2 | 6 |
| rs3812132 | chr6 | 87970301 | Synonymous | 1.08E-09 | ZNF292 | m6A_ID_195504 | 87970328 | Loss | 2 | 6 |
| rs3813250 | chr2 | 179395958 | Synonymous | 2.53E-15 | TTN | m6A_ID_147095 | 179395966 | Loss | 0 | 4 |
| rs383692 | chr6 | 88051444 | 3′-UTR | 1.86E-08 | SMIM8 | m6A_ID_195543 | 88051446 | Gain | 7 | 18 |
| rs4707358 | chr6 | 87994572 | Nonsynonymous | 6.11E-09 | GJB7 | m6A_ID_195538 | 87994573 | Loss | 0 | 9 |
| rs4894029 | chr2 | 179447848 | Synonymous | 8.35E-15 | TTN | m6A_ID_147800 | 179447846 | Loss | 0 | 4 |
| rs4963770 | chr12 | 24720617 | Exon | 3.08E-10 | LINC00477 | m6A_ID_258424 | 24720616 | Loss | – | – |
| rs7729926 | chr5 | 137514641 | Intron | 5.88E-16 | KIF20A | m6A_ID_183887 | 137514641 | Loss | 0 | 5 |
| rs774854 | chr3 | 111688578 | Synonymous | 9.80E-11 | PHLDB2 | m6A_ID_161546 | 111688577 | Loss | 0 | 1 |

In silico Replication of the m6A-SNPs
We randomly selected 22,982 non-m6A-SNPs from the AF GWAS dataset and calculated the number of SNPs with GWAS P-values<0.05. This randomization process was repeated 1000 times. On the basis of the 1000 randomizations, the proportion of AF associated m6A-SNPs with a GWAS P-value<0.05 was significantly higher than that of non-m6A-SNPs (95% CI: [5.76%, 5.78%], P-value<2.2×10−16). For the 1429 m6A-SNPs nominally associated with AF, we also performed an in silico replication by using a multi-ethnic GWAS dataset for AF in 2018, which contained fewer SNPs but more AF samples than the AF2018 dataset [15]. A total of 623 unique SNPs were nominally (P-value<0.05) associated with AF, and 86 m6A-SNPs overlapped with those from the AF2018 dataset.
A SNP is defined as an m6A-SNP if it has the potential to alter the DRACH motif [22] (where D=A, G or U; R=A or G; H=A, C or U) or other sequence features essential for m6A modification [14]. When examining the sequences around m6A sites for the 17 m6A-SNPs with genome-wide significant associations with AF, we observed that ten SNPs were located in the DRACH motif (Figure 4A). Notably, five SNPs (rs2846675, rs3211105, rs4707358, rs7729926 and rs774854) altered the core AC motif (Figure 4B).

Identification of 13 m6A-SNPs Showing eQTL Signals
To investigate the possible functional mechanisms underlying the 17 identified m6A-SNPs with genome-wide significant associations with AF, we tested their association with gene expression levels by using HaploReg. In total, 13 of the 17 AF-associated m6A-SNPs showed eQTL signals in various tissues or cells (Table 1; details in Table S1). Six m6A-SNPs displayed strong cis-eQTL signals with the gene being located, whereas the remaining seven m6A-SNPs showed only trans-eQTL signals. In particular, rs383692 (SMIM8) had strong cis-eQTL signals with SMIM8 in seven different tissues (Table S1). rs3211105 and rs1061259 in JMJD1C were found to be associated with JMJD1C expression in the testis, with P-values of 3.63×10−6 and 1.51×10−5, respectively. rs1152582 (SYNE2) was associated with SYNE2 expression in the left ventricle, with a P-value of 3.37×10−8.
Differential Expression Analysis
For the 13 m6A-SNPs displaying eQTL signals, we assessed the expression levels of their associated genes in three expression profile datasets (GSE14975, GSE108660 and GSE2240). After removing genes that were not detected on the microarray, we finally performed differential expression analysis for 19 associated genes (Table S2). Four genes, SYNE2, SMIM8, ZNF292 and JMJD1C-AS1, were differentially expressed in at least one of the three datasets. The gene SYNE2 was significantly down-regulated in AF samples compared with sinus rhythm samples in all three datasets (Figure 5A). Furthermore, the gene SMIM8 was significantly downregulated in two of the three datasets (Figure 5B). According to the eQTL results from HaploReg, the expression levels of SYNE2 were associated with m6A-SNP rs1152582 (cis-eQTL, P-value=3.37×10−8). The expression level of SMIM8 was associated with rs383692 (cis-eQTL), rs4707358 (trans-eQTL) and rs3812132 (trans-eQTL).
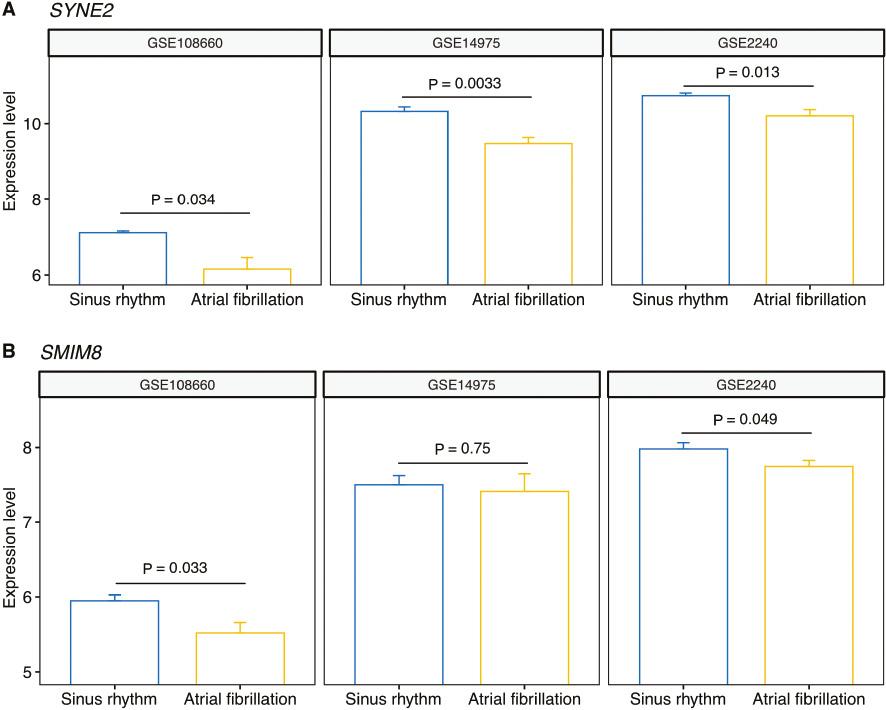
Expression Patterns of the Genes SYNE2 and SMIM8 in the AF and Sinus Rhythm Samples From the Datasets GSE14975, GSE108660 and GSE2240.
(A) The expression of SYNE2 was significantly downregulated in the AF group compared with the sinus rhythm group in all three datasets. (B) The expression of SMIM8 was significantly downregulated in the AF group compared with the sinus rhythm group according to the GSE108660 and GSE2240 datasets.
Discussion
Despite recent advances in GWAS data that have identified many AF-associated loci, methods to explore their functional implications have been less explored. To date, no genetic or experimental evidence has demonstrated an association between m6A RNA methylation and AF. The detection of m6A-SNPs enables the potential function of a subset of SNPs to be interpreted from the perspective of RNA methylation. In this study, we report the first exploration of the relationship between m6A-SNPs and AF through mining data from a large-scale GWAS. We identified 1429 m6A-SNPs associated with AF, among which 17 unique m6A-SNPs reached genome-wide significance. Among these AF-associated SNPs, five m6A-SNPs showed strong cis-eQTL signals with the genes in which they were located. Differential expression analysis indicated that SYNE2 was significantly down-regulated in AF. We hypothesized that m6A methylation modifications might participate in AF development by regulating SYNE2 expression. Our study provides population genetic evidence suggesting that m6A modifications are likely to be involved in the development of AF and to be associated with AF susceptibility.
Appropriate m6A RNA modification relies on the precise recognition and binding of RNA to methylation enzymes. SNPs influence RNA modification by affecting RNA secondary structures, RNA-protein interactions and exon splicing [23–25]. In particular, m6A is affected by SNPs occurring in the specific motif of the target RNA or the key flanking nucleotides [26]. Here, we identified 17 AF-associated m6A-SNPs in 14 genes (JMJD1C, JMJD1C-AS1, KANSL1, Pir-50760, SYNE2, MYO18B, TTN, KCNJ5, ZNF292, SMIM8, GJB7, LINC00477, KIF20A and PHLDB2). Among these genes, seven (SYNE2 [13, 15], MYO18B [13, 15], TTN [27], KCNJ5 [13, 15, 28], ZNF292 [15], LINC00477 [15] and PHLDB2 [15]) have been reported to be associated with AF in previous studies, whereas JMJD1C, JMJD1C-AS1, Pir-50760, KANSL1, SMIM8, GJB7 and KIF20A have not yet been reported. According to the Bgee database (https://bgee.org) [29], these genes are expressed in heart tissue. However, the functions of these genes in the heart, particularly in AF, remain to be further explored. The results of our study suggest novel potential links between each of the seven genes (JMJD1C, JMJD1C-AS1, Pir-50760, KANSL1, SMIM8, GJB7 and KIF20A) and AF development. Additionally, m6A RNA methylation may be the underlying mechanism.
Recent transcriptome-wide m6A maps suggest that m6A modification is restricted predominantly to adenosines in the DRACH motif [30, 31]. We compared the ten m6A-SNPs containing motifs to human assembly GRCh37 reference sequences and found that “AC” nucleotides were relatively evolutionarily conserved. This finding indicated that “AC” nucleotides in the m6A motif play a crucial role in m6A modification, in agreement with findings from previous studies [9, 22]. In addition, five of ten m6A-SNPs occurred in “AC” nucleotides, thereby suggesting that variants in “AC” in the m6A motif may substantially affect the physiological functions of m6A modification.
The novel m6A motifs derived by SNPs have a higher frequency than matched control alleles or neutral sites, thus suggesting that some of these SNPs are subject to positive selection [32]. We observed no significant difference in allele frequency between the loss-of-function m6A-SNPs and reference alleles, thus suggesting that AF-associated m6A-SNPs did not have a selection trend in the population. This result might have been be partly due to the limited number of variants that we identified. Our eQTL analysis revealed that the AF-associated m6A-SNP rs1152582 was highly correlated with the expression level of SYNE2 (cis-eQTL). The SNP rs1152582 is located in the 3’-UTR of the SYNE2 gene, in agreement with previous observations that most m6A site enrichment occurs in 3´-terminal exons and the 3’-UTR [22]. m6A modification regulates mRNA stability and target gene expression. We further used three independent GEO datasets to confirm that SYNE2 expression was significantly diminished in the AF group, thus suggesting that differences in SYNE2 expression may contribute to AF development and that m6A methylation modifications may be involved in the regulation of SYNE2 expression.
The SYNE2 gene is ubiquitously expressed and consists of 116 exons that encode the Nesprin-2 protein. Alternative splicing of gene produces multiple isoform transcripts with tissue-specific expression patterns [33]. Notably, Nesprin-2α1, a small isoform encoded by exons 106–116, is highly expressed in cardiac and skeletal muscle cells [33]. Nesprin-2, located mainly in the outer nuclear membrane of the nuclear envelope, contains two major domains: the N-terminal actin-binding domain and the C-terminal Klarsicht/Anc-1/Syne homology (KASH) domain [34]. Nesprin-2 functions as a scaffold linking the nucleoskeleton and cytoskeleton, and forms part of the LINC complex [35]. Interactions with various partners, such as F-actin, SUN2, Lamin A/C and Emerin, regulate Nesprin-2’s functions [33, 36, 37]. In cardiomyocytes, Nesprin-2 is crucial for maintaining nuclear morphology, positioning, mechanotransduction and nuclear trafficking [38–41]. Knocking out Nesprin-1/2 has been shown to cause early-onset cardiomyopathy and cardiac dysfunction [41]. These results suggest that SYNE2 plays a critical role in cardiac physiology. However, further investigations are necessary to determine the additional functions of this gene.
Conclusion
In conclusion, our study identified several m6A-SNPs that may contribute to the genetic risk of AF, thus highlighting the potential role of m6A modification in AF pathogenesis. Our findings also emphasize the crucial role of SYNE2 in AF. However, the lack of in vivo experimental verification of the effects of genetic variations on m6A modification in candidate transcripts and their subsequent functions in the pathogenesis of AF is a substantial limitation of this study. Future studies should focus on functional experimental verification to investigate the specific mechanisms involved in these genetic contributions. Comprehensive elucidation of the relationship between m6A modification and AF would deepen understanding of AF pathogenesis and potentially lead to the development of targeted therapies for this disease.