
Significance statement: Many challenges and difficulties exist in the treatment of HFpEF, owing to clinical heterogeneity. Recently, SGLT2 inhibitors have been found to confer clinical benefits for HFpEF, but the specific mechanisms remain unclear. This research demonstrated that CANA ameliorated heart function in HFpEF rats, partly by regulating ferroptosis, potentially through via AMPK/PGC-1α/Nrf2 signaling. Our data may provide novel insights into the mechanisms of SGLT2 inhibitors, and new targets for HFpEF prevention and treatment.
Introduction
Heart failure with preserved ejection fraction (HFpEF) is currently a public health problem in the cardiovascular field because of its high prevalence, accounting for more than half of patients with heart failure (HF), and progressive increase in mortality [1]. Several clinical trials have shown that conventional drugs for heart failure with reduced ejection fraction (HFrEF) are not effective in patients with HFpEF [2]. Unexpectedly, the results of the EMPEROR-Preserved study in 2021 and the DELIVER study in 2022 showed that empagliflozin and dapagliflozin significantly decreased the incidence of primary composite endpoint events (cardiovascular death and hospitalization due to HF) in patients with HFpEF, regardless of the presence of diabetes [3, 4]. Empagliflozin and dapagliflozin, a new class of hypoglycemic agents, are SGLT2 inhibitors, which act mainly in the kidneys and exert hypoglycemic effects by inhibiting urinary glucose reabsorption [5]. This class of drugs has long been of interest because of their cardiovascular benefits [6, 7], but the exact mechanism of action has been unclear. Ferroptosis is a form of programmed cellular necrosis caused by iron-dependent lipid peroxidation accompanied by reactive oxygen species overload [8]. Studies have confirmed that ferroptosis is involved in the pathophysiology and evolution of cardiovascular diseases, including HF [9]. Adenosine monophosphate activated protein kinase (AMPK) regulates metabolism and biosynthesis through peroxisome proliferator-activated receptor gamma coactivator-1-α (PGC-1α) [10], which strongly induces nuclear factor E2 related factor 2 (Nrf2). Nrf2 and HO-1 are downstream mediators in this pathway, which regulate proteins involved in ferroptosis [11, 12]. However, HO-1 both positively and negatively regulates ferroptosis [13]. Therefore, in this study, to provide novel insights into the cardioprotective benefits of SGLT2 inhibitors, we explored whether AMPK/PGC-1α/Nrf2 signaling might be activated by CANA and act in ferroptosis regulation.
Materials and Methods
Animal Model
As described in our previous study [14], 36 specific-pathogen-free male Dahl salt-sensitive (DSS) rats were randomly divided into three groups: (1) a control group (normal group) receiving a low-salt diet (0.3% NaCl+AIN-76a) and 0.5% hydroxypropyl methylcellulose (HPMC) (2 mL/kg/d) by gavage; (2) an HFpEF group receiving a high salt diet (8% NaCl+AIN-76a) and 0.5% HMPC (5 mL/kg/d) by gavage; and (3) an HFpEF group with CANA (CANA group) receiving a high-salt diet (8% NaCl+AIN-76a) and CANA (20 mg/kg/d) dissolved in 0.5% HMPC (2 mL/kg/d) by gavage. The rats were observed for 12 weeks.
Blood Pressure Measurements
Rats were placed into the sleeve and fixed on the tail-cuff plethysmography (BP-2000; Visitech Systems, Inc.). When they stopped agitating and the pulse was relatively stable, the blood pressure was measured for 15 min at weeks 0, 4, 8 and 12. Six valid blood pressure values were averaged, and the mean arterial pressure was calculated.
Echocardiography
In the 12th week of the experiment, six DSS rats were randomly selected in each group for cardiac echocardiography with a Vevo2100 with high-resolution small animal echocardiograph with an MS400 high-frequency probe (ultrasound frequency range 18–38 MHz). After the rats had been skinned and anesthetized by isoflurane inhalation, indexes were measured by acquiring long-axis views of the left ventricle and apical four-chamber views. Echocardiography was performed by an experienced sonographer who was blinded to the experimental groups. After three consecutive cardiac cycles, the mean value was calculated.
Experimental Specimen Collection
At the end of the 12th week, after anesthesia, the rats were euthanized by blood collection from the abdominal aorta. Hearts were removed and placed in 4°C PBS solution. After the residual blood was washed away, and the surface was blotted with filter paper, the left ventricular tissue was cut into small pieces, placed in liquid nitrogen and transferred to a −80°C freezer for detection. In addition, some tissues were fixed in 4% paraformaldehyde solution and then embedded in paraffin for pathological examination.
Hematoxylin and Eosin (HE) Staining
Paraffin sections were sealed after successive steps of dewaxing, washing with water, hematoxylin staining, hydrochloric acid ethanol separation, gradient ethanol solution treatment, eosin solution staining, dehydration and dewaxing. Cardiomyocyte morphology was observed under ordinary light microscopy and photographed, and the cross-sectional area (CSA) was measured in Image-Pro Plus 6.0.
Masson Staining
Paraffin sections were sealed after dewaxing, washing, staining with Weigert hematoxylin mixture and Ponceau acid fuchsin solution, soaking in 1% phosphotungstic acid solution, staining with aniline blue solution, gradient alcohol dehydration and dewaxing. The sections were microscopically examined and photographed under an ordinary light microscope, and the collagen volume fraction (CVF) was determined in Image-Pro Plus 6.0.
Prussian Blue Iron Staining
Paraffin sections were placed in Perls staining solution and Karyfast red staining solution successively, then dehydrated, sealed, examined under a light microscope and photographed.
Protein Expression Detection
Western blotting was performed to detect the expression of AMPK (Affinity Biosciences, USA), P-AMPK (Affinity Biosciences, USA), PGC-1α (Abcam, UK), Nrf2 (Proteintech, Wuhan, China), HO-1 (Proteintech, Wuhan, China), GPX4 (Abcam, Cambridge, UK), ACSL4 (Abcam, Cambridge, UK) and FTH1 (Abcam, Cambridge, UK) in rat myocardial tissue. A BCA protein concentration assay kit (Solebro, Beijing, China) was used to determine the protein concentrations of the samples. After protein denaturation, gel preparation, sample loading, electrophoresis, gel cutting, membrane transfer, blocking and antibody incubation, the bands were developed and imaged. The grayscale values of the bands were analyzed in ImageJ.
mRNA Expression Detection
The expression of PGC-1α, Nrf2 and HO-1 mRNA in rat myocardial tissue was detected by reverse transcription–quantitative polymerase chain reaction (RT-qPCR) (Table 1). Approximately 30 mg of myocardial tissue was collected and added to 1 mL of TRNzol Universal reagent (Tiangen Biochemical Technology Co., Ltd., Beijing, China). RNA was extracted after samples were thoroughly homogenized. The optical density values were measured with an ultraviolet spectrophotometer at 260 nm and 280 nm, and the ratio of measurements was calculated. If the ratio was between 1.8 and 2.0, the total RNA purity was considered adequate. A gDNA removal reaction system (Tiangen Biochemical Technology Co., Ltd., Beijing, China) and reverse transcription reaction system (Tiangen Biochemical Technology Co., Ltd., Beijing, China) were used. The RT-qPCR reaction system was prepared, and PCR was performed. The relative quantity value was calculated with the 2-ΔΔCt calculation method to quantify the expression levels of target genes.
Primer Sequences for RT-qPCR Target Genes.
| Target gene | Forward sequence (5′-3′) | Reverse sequence (5′-3′) | Sequence length |
|---|---|---|---|
| PGC-1α | AGAACCATGCAAACCACACC | TTGGTGTGAGGAGGGTCATC | 172 |
| Nrf2 | TTGGCAGAGACATTCCCATTTGTA | GAGCTATCGAGTGACTGAGCCTG | 109 |
| HO-1 | GAGCAGAGCCATCTCGAGCG | ACCTCCTTGGTGGCCTCCTT | 143 |
| β-actin | CACCATGTACCCAGGCATTG | CCTGCTTGCTGATCCACATC | 20 |
Statistical Analysis
SPSS 26.0 was applied to analyze the data. Normally distributed measures are expressed as mean±SD. One-way ANOVA was used for comparisons among multiple groups. Bonferroni’s post hoc test was performed when the variances were equal. Otherwise, Tamhane’s post hoc test was used. Non-normally distributed measures are expressed as mean (Q1, Q3). The Kruskal-Wallis H rank sum test was used for comparisons between groups. P<0.05 was considered statistically significant.
Results
General Condition of DSS Rats
All 24 DSS rats in the normal and CANA groups survived until the end of the study and showed good mental and physical characteristics. Four DSS rats in the HFpEF group died in the 10th–12th weeks, and showed a significant loss of appetite, depression and diminished activity before death. No pathological autopsy was performed after death.
HFpEF Model Construction and Verification
The blood pressure in DSS rats in the HFpEF group gradually increased after feeding with a high-salt diet, and was persistently higher than those in the control and CANA groups (P<0.05) (Figure 1). At the 12th week, the HFpEF group showed lower E/A ratios (<1) than the control group (P<0.05). However, no significant changes were observed in left ventricular internal diameter in end diastole (LVIDd), left ventricular internal diameter in end systole (LVIDs), ejection fraction (EF) and fractional shortening (FS) (P>0.05) (Table 2). The myocardial pathology in the HFpEF group showed a disorganized myofibrillar arrangement, enlarged CSA (P<0.05), irregular morphology and interstitial collagen fiber hyperplasia with elevated CVF compared with Normal group (P<0.05) (Figure 2). The HFpEF model was successfully developed, on the basis of the characteristics described above.
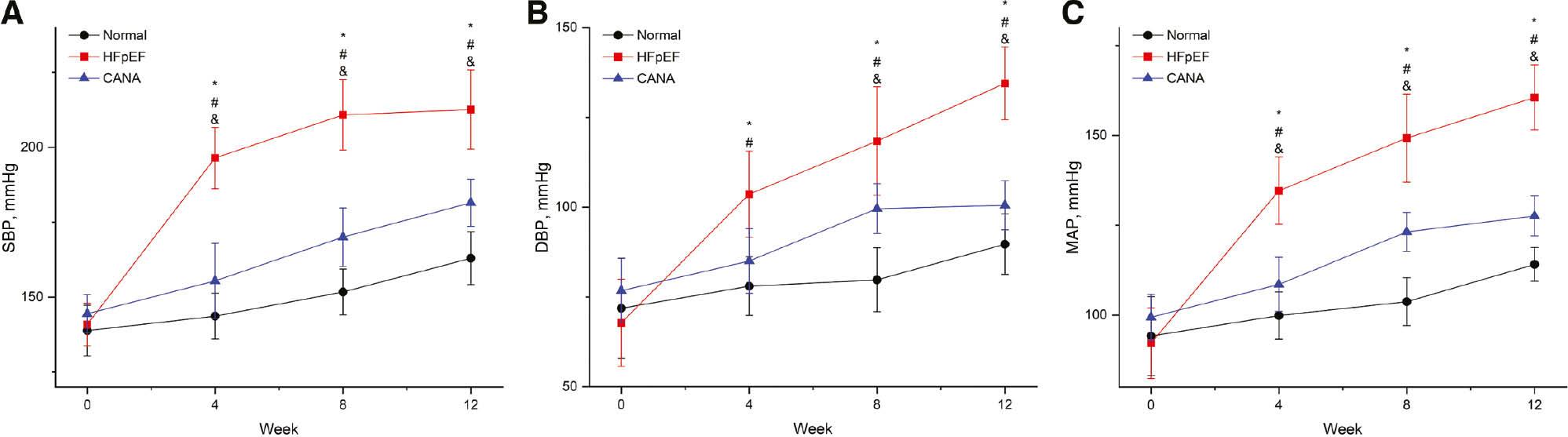
Blood Pressure Measurement in each Group.
(A) SBP, systolic blood pressure. (B) DBP, diastolic blood pressure. (C) MAP, mean arterial pressure. *P<0.05 HFpEF group versus normal group. #P<0.05 CANA group versus HFpEF group. &P<0.05 CANA group versus normal group.
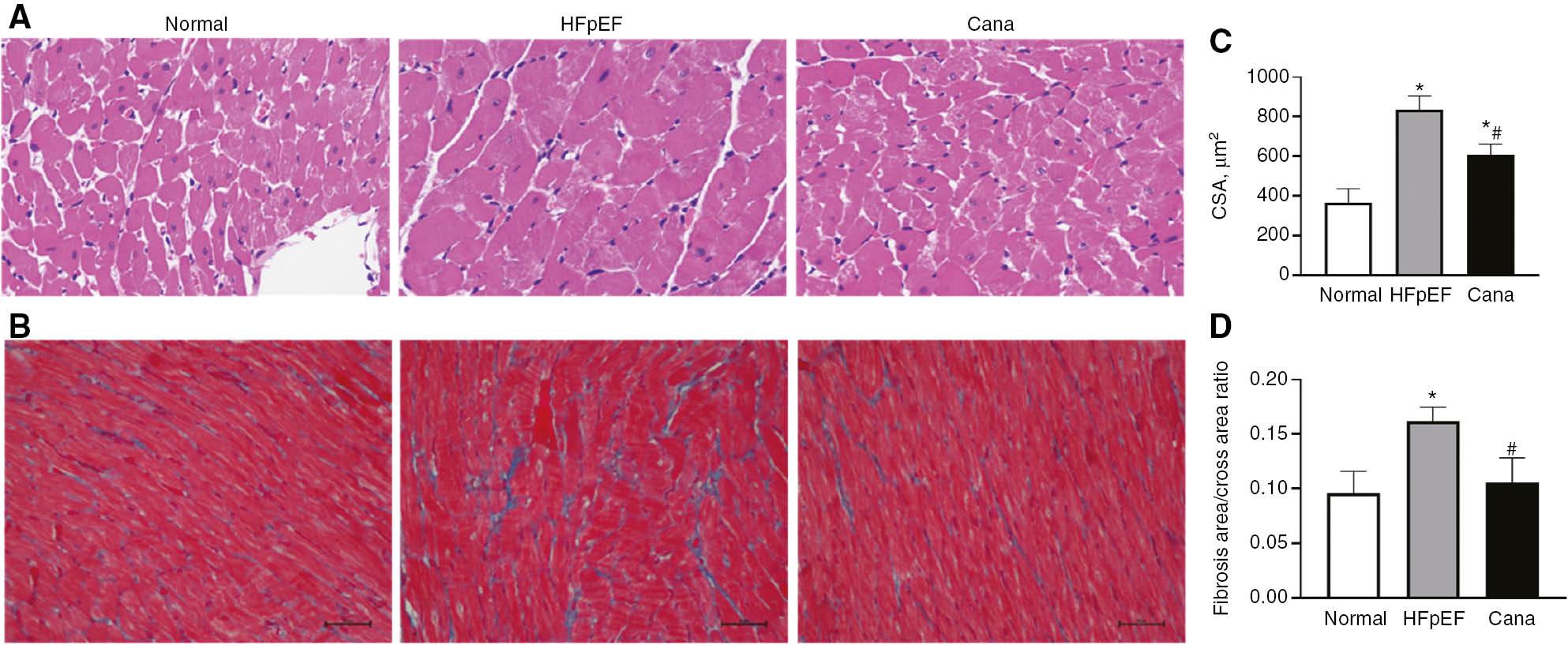
HE Staining and Masson Staining of Myocardial Tissue in each Group.
(A) HE staining graphs. (B) Masson staining graphs. (C) CSA comparison. (D) Interstitial fibrosis CVF comparison. *P<0.05 versus normal group. #P<0.05 versus HFpEF group.
Echocardiographic Assessment of Rats in each Group.
| Normal n=6 | HFpEF n=6 | Cana n=6 | P | |
|---|---|---|---|---|
| LVIDd (mm) | 7.29±0.34 | 7.22±0.23 | 7.08±0.48 | 0.603 |
| LVIDs (mm) | 3.41±0.12 | 3.69±0.27 | 3.53±0.45 | 0.311 |
| EF (%) | 80.54±5.29 | 80.60±4.84 | 77.06±4.08 | 0.363 |
| FS (%) | 53.23±2.31 | 48.73±4.88 | 49.11±3.34 | 0.094 |
| E/A | 1.24±0.11 | 0.91±0.05* | 1.15±0.13# | 0.0001 |
Data are presented as mean±standard deviation. *P<0.05 versus normal group. #P<0.05 versus HFpEF group. LVIDd, left ventricular internal diameter in end diastole; LVIDs, left ventricular internal diameter in end systole; EF, ejection fraction; FS, fractional shortening.
Effects of CANA on HFpEF Rats
The blood pressure in rats in the CANA group fed a high-salt diet and simultaneously administered CANA was higher than that in the control group but lower than that in the HFpEF group (P<0.05) (Figure 1), thus suggesting that CANA has an antihypertensive effect. The echocardiograms in the CANA group showed greater E/A ratios than those in the HFpEF group (P<0.05). LVIDd, LVIDs, EF and FS showed no significant changes among the three groups (P>0.05) (Table 2). Myocardial pathology was also ameliorated (Figure 2). These changes indicated that CANA improved left ventricular diastolic function in HFpEF rats.
Effects of CANA on Ferroptosis
The expression of key proteins involved in ferroptosis was assessed. The expression of ACSL4 was clearly elevated in HFpEF rats compared with the control group (P<0.05), whereas the expression of FTH1 was significantly diminished (P<0.05), and GPX4 did not markedly change (P>0.05). Prussian blue iron staining demonstrated that CANA ameliorated iron overload in HFpEF rats. Meanwhile, CANA reversed changes in ACSL4 and FTH1, but did not alter the expression of GPX4 (Figure 3). We deduced that ferroptosis occurred in HFpEF rats, and CANA regulated ferroptosis, thereby improving heart function.

Effects of CANA on the AMPK/PGC-1α/Nrf2 Signaling Pathway
No significant difference in AMPK expression was observed among groups. However, compared with those in the normal group, pAMPK and the pAMPK/AMPK ratio were lower in the HFpEF group (P<0.05) but significantly increased after CANA treatment (P<0.05). Western blotting and RT-qPCR indicated that the mRNA and protein levels of PGC-1α, Nrf2 and HO-1 in the HFpEF group were lower than those in the normal group, and then reverted after the CANA intervention (P<0.05) (Figure 3). We deduced that CANA regulates ferroptosis, possibly via activating AMPK/PGC-1α/Nrf2 signaling in HFpEF rats.
Discussion
With the increasing age of the population and the prevalence of chronic diseases such as obesity, diabetes and hypertension, HFpEF has become a common manifestation of HF worldwide. Many challenges and difficulties are faced in the treatment of HFpEF, owing to clinical heterogeneity. SGLT2 inhibitors, newly available hypoglycemic agents, have attracted attention for their substantial cardiovascular benefits. Supported by the results of the EMPEROR-Preserved study [3], this class of drugs has provided new opportunities for the treatment of HFpEF. Relevant basic science studies using HFpEF as a model have shown that SGLT2 inhibitors ameliorate myocardial inflammation, oxidative stress and vascular endothelial function; inhibit aortic sympathetic nerve activity; and decrease myocardial oxygen consumption and cardiac anterior and posterior loads in HFpEF experimental animals [15–17]. However, the specific mechanism is unclear. In this study, we demonstrated that CANA improved heart function in HFpEF rats, partly by regulating ferroptosis, which was potentially activated via AMPK/PGC-1α/Nrf2 signaling.
The concept of ferroptosis was first introduced internationally in 2012 by Dixon et al. [8]. This form of programmed cell necrosis involves iron metabolism, lipid metabolism, amino acid metabolism and other metabolic processes. Ferritin can chelate 4500 iron atoms, thereby protecting cells against free iron and maintaining iron homeostasis [18]. Polyunsaturated fatty acids in phospholipids of cell and organelle membranes are acylated through the action of ACSL4 [19], and esterification and oxidation subsequently generate lipid peroxidation products, which are converted to non-toxic fatty alcohols by glutathione under the catalysis of GPX4 [20]. Ferroptosis is activated in the presence of intracellular iron overload, excessive production of lipid peroxides or diminished GPX4 activity. GPX4, ACSL4 and FTH1 are key proteins in this process. Ferroptosis has been found to play a key role in the development of heart failure. Ferroptosis-associated protein changes have been found to be inhibited by overexpression of mTOR; inhibition of the JNK/p53 signaling pathway; and application of iron chelators such as desferrioxamine and dextropropylenimine, and drugs such as gerberoside and atorvastatin, thus improving cardiac function [21– 25]. In our study, in HFpEF rats, we observed iron accumulation, increased ACSL4 and decreased FTH1, thus indicating that ferroptosis was induced and then ameliorated by CANA. However, the expression of GPX4 did not differ among the three groups. We inferred that in the HFpEF model, decreased glutathione synthesis might be critical, rather than GPX4 inactivation. This inference had been verified in our previous research [14].
Unlike other cell death pathways, ferroptosis can be induced by both experimental small molecules (e.g., erastin, Ras lethal small molecules 3 and sulfadiazine) and certain drugs (e.g., lorazepam, sorafenib and artemisinin) [26, 27]. Furthermore, it can be inhibited by substances such as ferrostatins and liproxstatins. Moreover, the mechanism of ferroptosis is regulated. The positive regulatory biomolecules include voltage-dependent anion channel 2/3, Ras, nicotinamide adenine dinucleotide phosphate oxidase and p53. The negative regulatory biomolecules include glutathione peroxidase, solute carrier family 7-member 11, heat shock protein B1 and Nrf2 [26]. Therefore, all metabolic pathways affecting the expression of the above biomolecules accordingly regulate the ferroptosis mechanism.
AMPK is a well-known energy sensor and metabolic regulator that promotes fatty acid oxidation and inhibits lipid synthesis. It is a target in many disease mechanisms, because of its critical role in energy metabolism [28–30]. AMPK deficiency has been reported to induce cardiac systolic dysfunction and dilated cardiomyopathy [31]. AMPK not only regulates cellular metabolic status and mitochondrial biosynthesis by regulating PGC-1α [10], but also modulates oxidative stress directly or through the induction of Nrf2 by PGC-1α [32–34]. In response to oxidative stress, AMPK promotes the dissociation of Nrf2 from Kelch ECH-associated protein 1, interferes with Nrf2 ubiquitination, and causes nuclear translocation of Nrf2. Moreover, PGC-1α is translocated into the nucleus, where it upregulates Nrf2 expression by binding peroxisome proliferator-response elements in the promoter of Nrf2 [35]. Subsequently, the up-regulation of Nrf2 promotes the transcription of antioxidant response element-mediated genes [36] (Figure 4). Through scrutiny of these genes, we concluded that Nrf2 is one of the most important transcription factors modulating cellular oxidative stress, as well as a key factor in maintaining cellular redox homeostasis. Nrf2 mediates the constitutive and inducible expression of antioxidant proteins of a series of defense systems, such as quinone oxidoreductase 1 and HO-1, thus attenuating cellular damage induced by reactive oxygen species and electrophiles. HO-1 degrades heme iron into Fe2+, heparin and carbon monoxide, thereby increasing iron load; simultaneously, it is an antioxidant that attenuates oxidative stress. Thus, it has dual regulatory roles on ferroptosis mechanisms [13]. Our research confirmed that the antioxidant effect of HO-1 may exceed its heme iron degradation in the HFpEF model with CANA intervention. Nrf2 also modulates ferroptosis mechanisms by transcriptionally regulating key proteins involved in iron metabolism [12] and almost all genes involved in ferroptosis, including glutathione-regulated genes, reducing nicotinamide adenine dinucleotide phosphate regeneration genes that are critical for glutathione peroxidase 4 activity, and lipid peroxidation and iron metabolism related genes [37, 38]. Thus, the AMPK/PGC-1α/Nrf2 signaling pathway regulates the ferroptosis process. Our previous proteomic analysis has indicated that the PPAR signaling pathway is a significantly enriched KEGG metabolic pathway between the HFpEF and CANA groups [14], and the AMPK/PGC-1α/Nrf2 pathway is an important part of the PPAR signaling pathway. Therefore, we proposed and tested the hypothesis that the mechanism of ferroptosis regulation by CANA might be based on the AMPK/PGC-1α/Nrf2 signaling pathway. The results indicated that this signaling pathway was activated by CANA, thus supporting our hypothesis.

On the basis of our experiments, we suggest that CANA may regulate the ferroptosis mechanism in HFpEF rats through the AMPK/PGC-1α/Nrf2 signaling pathway, thus improving cardiac function. These findings may provide new clues regarding the mechanism underlying the cardiovascular benefits of SGLT2 inhibitors. However, the upstream and downstream relationships cannot be demonstrated at present without the application of ferroptosis-associated inducers or blockers, as well as gene knockdown techniques to corroborate this signaling pathway. In future studies, we intend to complement our findings by applying cellular experiments through construction of a drug-induced HF cardiomyocyte injury model to further validate the reliability of the upstream pathway. Therefore, further exploration of the detailed molecular mechanism of CANA regulation of ferroptosis is necessary.
In conclusion, canagliflozin regulates ferroptosis, potentially via activating AMPK/PGC-1α/Nrf2 signaling in HFpEF rats.