
Introduction
Titin (TTN) is the largest human protein, with a molecular mass of 4200 kDa (38,138 amino acid residues) [1], and the third most abundant protein in the heart [2]. TTN contains four regions: the Z disk, I band, A band, and M line regions [3]. Mutations in the TTN gene can result in many cardiomyopathies, including hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM). HCM is the leading cause of unexpected cardiac death each year [4], while DCM 5-year mortality is 15–50% [5]. Many DCM patients experience cardiac arrhythmia, heart failure, and stroke [6]. In China, TTN mutations resulting in DCM are often reported. Lyu et al. [7] reported that a TTN missense mutation (c.100126A>G/p.Thr33376Ala) is responsible for familial DCM. Another report also observed an association between TTN missense mutation and DCM risk in Jiangsu, China [8]. Liu et al. [9] subsequently reported two DCM families harboring a nonsense mutation (c.12325C>T/p.Arg4109Ter) and a missense mutation (c.17755G>C/p.Gly5919Arg). It appears that missense mutations in Chinese DCM patients are common. Given that there are thousands of amino acid residues in the protein, the mutations already reported cannot represent the whole spectrum of TTN mutations. To further identify the DCM-causing gene spectrum in China, we assessed three familial DCM pedigrees (see Figure 1) and studied 101 genes (see Supplement 1) associated with cardiomyopathy by whole exome sequencing and Sanger sequencing. We found that each family harbors a mutation (nonsense or splice site mutation) in TTN, which is likely the cause of their disease. All the mutations had not been reported previously. At the same time, we collected detailed clinical data for each family, and confirmed them with the sequencing results, thus providing further clues to explore the DCM mechanism.

Materials and Methods
Ethics Statement
This study was conducted with the approval of the Ethics Committee of the First Affiliated Hospital of Zhengzhou University, China. Informed consent forms were signed by study participants before blood collection. This study was conducted under the guidelines of the Declaration of Helsinki.
DNA Extraction and High-Throughput Genome Screening
Blood (10 mL) was collected from participants and stored at 4°C. Peripheral blood mononuclear cells (PBMCs) were separated from the peripheral blood, and DNA was extracted from the PBMCs with Ficoll-Paque PLUS (density 1.077 mg/L) (GE Healthcare, USA). Next-generation sequencing (NGS) was performed to detect single base pair mutations and/or small fragment deletions or insertions in the cardiomyopathy-related genes at the Zhengzhou Clinical Medical Laboratory (Zhengzhou, China). Sequencing was conducted by an individual blinded to the clinical features of the study participants. The NGS was performed with a NextSeq CN500 sequencer (Berry Genomics Corporation), and Agilent SureSelect Human All Exon V6 kits were used to create the exome library on the Illumina HiSeq X-10 platform. Sanger sequencing was performed for further confirmation of the precise mutation location. The polymerase chain reaction (PCR) primers were designed with PRIMER 6.0, and the PCR programs were run on a 3100 Genetic Analyzer (ABI, Foster City, CA, USA). The PCR conditions were as follows: initial denaturation at 95°C for 5 minutes; 35 cycles of denaturation at 95°C for 30 seconds, annealing at 59°C for 30 seconds, and extension at 72°C for 55 seconds; and a final extension at 72°C for 15 minutes, followed by a hold at 4°C until sequencing. The exact PCR primers for the three families are given in Table 1.
Clinical Data Collection and Evaluation
Every proband’s family and medical history were collected. A physical examination was performed by physicians at the First Affiliated Hospital of Zhengzhou University, and probands were questioned about their life habits and customs. An electrocardiogram (ECG), B-mode echocardiography, and cardiac magnetic resonance imaging (MRI) were performed for all of the probands and some of their relatives.
Results
Identification of the TTN Mutation
Genomic DNA was isolated from PBMCs in the peripheral blood of the families. We focused on genes that have been associated with cardiomyopathy and sequenced their whole exomes by NGS. Subsequently, Sanger sequencing was performed to identify the precise mutation in the TTN gene.
We found that each of the probands assessed inherited a heterozygous mutation in the TTN gene. All the mutations can influence TTN production and folding. Figure 2 reveals that some of their relatives share the same mutation in the three families. We searched for the mutation in dbSNP (https://www.ncbi.nlm.nih.gov/snp/), the 1000 Genomes Project database (http://www.internationalgenome.org/), the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/), and the ExAC database (http://exac.broadinstitute.org/) with a global minor allele frequency of 5% or greater. We could not find the three mutations (c.G20137T,c. G52522T,c.44610-2A>C) in the three families in any of the databases. The mutation information and protein alterations are summarized in Table 2. We also noted that there are some other gene mutations in the probands but most of them are variations of unknown significance; they are included in Table 3.
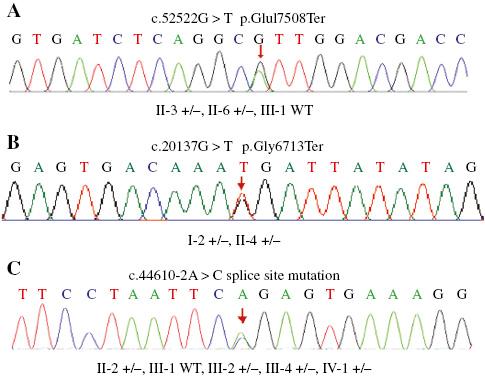
Sequence Results for the Family Pedigrees.
All individuals who were recruited for sequencing are shown. Participants for whom sequencing was not done were either rejected for this study or were unavailable. WT, wild type.
Variants in the TTN Gene in Three Pedigrees with Dilated Cardiomyopathy.
| Family | Nucleotide change | Mutation results | Previous publication | ACMG level |
|---|---|---|---|---|
| A | c.52522G>T | p.Glu17508Ter | No | Likely pathogenic |
| B | c.20137G>T | p.Gly6713Ter | No | Likely pathogenic |
| C | c.44610−2A>C | Splice site mutation | No | Likely pathogenic |
ACMG, American College of Medical Genetics and Genomics.
Other Variants Detected in the Probands.
| Proband | Gene | Nucleotide change | Mutation results | ACMG level |
|---|---|---|---|---|
| Family A | TTN: NM_133378 | c.36987G>A | p.Lys12329= | Likely benign |
| SYNE1: NM_033071 | c.11038−2A>G | Splice site mutation | Likely pathogenic | |
| Family B | MYH7: NM_000257 | c.4239G>A | p.Ser1413= | Likely benign |
| DMD: NM_004006 | c.10619C>T | p.Pro3540Leu | VUS | |
| DSG2: NM_001943 | c.2959G>T | p.Val987Phe | VUS | |
| ELAC2: NM_018127 | c.1274A>G | p.Tyr425Cys | VUS | |
| DYSF: NM_003494 | c.2934G>C | p.Glu978Asp | VUS | |
| SOS1: NM_005633 | c.1770G>A | p.Glu590= | Likely benign | |
| Family C | TGFB3: NM_003239 | c.288C>T | p.Thr96= | Likely benign |
| SYNE1: NM_033071 | c.12401C>T | p.Ser4134Leu | VUS | |
| TFR2: NM_003227 | c.1090G>A | p.Ala364Thr | VUS | |
| PFKM: NM_001166687.1 | c.−9+4G>T | Splice site mutation | VUS | |
| SPEG: NM_005876 | c.2597A>G | p.Lys866Arg | VUS | |
| TFR2: NM_003227 | c.1090G>A | p.Ala364Thr | VUS | |
| ILK: NM_004517 | c.707A>G | p.Asn236Ser | VUS | |
| OPA1: NM_015560 | c.565G>A | p.Glu189Lys | VUS | |
| TRDN: NM_006073 | c.1313T>A | p.Ile438Asn | Benign |
ACMG, American College of Medical Genetics and Genomics; VUS, variation of unknown significance.
Clinical Data
The main clinical features of the probands in the three families studied are shown in Table 4. In family A, two members harbored the TTN exon 154 c.52522G>T mutation. The proband in familyA (II-3) was a 53-year-old woman in whom diabetes had been diagnosed 8 years previously and in whom a dilated left ventricle had been found 5 years previously. In the previous 8 years, she had experienced cerebral stroke three times. In the previous year, the patient’s cardiac performance was greatly impaired, with a low left ventricular ejection fraction (LVEF) of 24.07% and a septum thickness of 6 mm as revealed by cardiac MRI. Cardiac MRI revealed some anomalous signals in the free wall and apex that profoundly indicated the noncompaction of the ventricular myocardium. Whole heart dilatation with a New York Heart Association (NYHA) function class of III had recently been diagnosed. A 24-hour dynamic ECG showed her average heart beat was approximately 91 per minute with serious ventricular tachycardia and the ST segment was changing dynamically. Individual I-1 of familyA died at the age of 72 years, 8 years previously, of heart failure and whole heart dilatation with poor cardiac function. He also had diabetes for many years. We did not have access to his blood, and his DNA sequence is unknown. Individual II-6 (47 years old) of familyA is the younger sister of the proband and often experiences palpitations and feels flustered. ECG led to a diagnosis of frequent premature ventricular contraction and paroxysmal ventricular tachycardia. She also had high blood glucose content, but diabetes was not diagnosed. Individual I-1 of familyA, the father of the proband, died at the age of 73 years before we could collect his blood. He died of serious heart failure accompanied by overall heart dilatation and diabetes. There are no obvious abnormalities in the other family members at the time of our research.
Clinical Data for the Three Families.
| Individual | Age/sex | Clinical features | Ultrasonography | ECG | Cardiac MRI | Genotype | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| EF (%) | LVDD (mm) | IVST (mm) | PW (mm) | FS (%) | EDV (mL) | ESV (mL) | |||||
| Family A | |||||||||||
| I-1 | 72/M | DCM, HF, diabetes | NA | NA | NA | +/− | |||||
| II-3 | 53/F | DCM, HF, diabetes | 24 | 54 | 8 | 9 | 17 | NSR | 177 | 134 | +/− |
| II-6 | 47/F | Palpitation, diabetes | 65 | 46 | 10 | 10 | 47 | FPVC, VT | NA | +/− | |
| Family B | |||||||||||
| I-2 | 57/F | DCM, edema | 30 | 54 | 10 | 10 | 14 | NSR | NA | +/− | |
| II-4 | 32/M | DCM | 35 | 66 | 10 | 11 | 17 | LBBB | 362 | 305 | +/− |
| Family C | |||||||||||
| II-2 | 62/F | DCM, dyspnea | 39 | 74 | 11 | 10 | 19 | PAF, FPVC | 381 | 313 | +/− |
| II-4 | 59/M | DCM, diabetes | 27 | 74 | 9 | 9 | 13 | L-QT | NA | NA | |
| III-1 | 37/M | Normal | 62 | 45 | 8 | 9 | 33 | NSR | NA | WT | |
| III-2 | 32/F | LV dilation | 67 | 56 | 9 | 8 | 37 | NSR | NA | +/− | |
| III-4 | 26/M | DCM, LVNC | 37 | 72 | 9 | 9 | 18 | NA | 419 | 360 | +/− |
| IV-1 | 8/M | Normal | NA | NA | NA | +/− | |||||
DCM, dilated cardiomyopathy; ECG, electrocardiogram; EF, ejection fraction; EDV, end-diastolic volume; ESV, end-systolic volume; F, female, FPVC, frequent premature ventricular contraction; FS, fractional shorting; HF, heart failure; IVST, end-diastolic interventricular septal thickness; LBBB, left bundle branch block; L-QT, long QT interval; LV, left ventricular; LVDD, left ventricular diastolic diameter; LVNC, left ventricular noncompaction; M, male; MRI, cardiac magnetic resonance imaging; NA, not available; NSR, normal standard electrocardiogram; PAF, persistent atrial fibrillation; PW, posterior left ventricular wall thickness; VT, ventricular tachycardia; WT, wild type; +/−; heterozygous.
In family B, two individuals (I-2 and II-4) were found to be carrying the TTN exon 79 c.20137G>T mutation. Since all third-generation individuals were younger than 10 years, their blood samples were not collected. The proband (I-2) was a 56-year-old woman in whom DCM had been diagnosed 4 years previously because of edema (especially of the face), nausea, and emesis. She had recently been hospitalized because of heart failure with LVEF of about 30% and fractional shortening (FS) of 14%. Further biochemical examination indicated serious electrolyte disturbance, and her aspartate aminotransferase level was 66 U/L and her pro-B-type natriuretic peptide level was 264.17 pg/mL. DCM had been diagnosed in her 32-year-old son (II-4) 3 years previously. B-mode ultrasonography demonstrated he had impaired left ventricular function with LVEF of 35% and FS of 17%. An ECG showed a left bundle branch block, a depressed ST segment, and an inverted T wave. He received a diagnosis of digestive tract edema with cardiac function class III (NYHA). We did not obtain blood samples from other family members, and they had no obvious cardiomyopathy symptoms .
In family C, individuals II-2, III-2, and III-4 were found to be harboring the heterozygous splice site mutation c.44610−2A>C. The proband (II-2) was a 62-year-old woman who had experienced chest tightness and shortness of breath for more than 8 years and in whom DCM had been diagnosed 4 years previously. An ECG and a 24-hour dynamic EGC showed abnormal but persistent atrial fibrillation, persistent ventricular tachycardia, and a continuous change in the ST-T segment. Cardiac ultrasonography demonstrated cardiac dilatation with a left ventricular end-diastolic diameter of 74 mm, LVEF of 39%, and FS of 19%. Biochemical examination showed an extremely high pro-B-type natriuretic peptide level of about 28,123 pg/mL, with additional electrolyte disturbance. Individual III-1 was 37 years old and her first son does not carry the TTN mutation. Individual III-2 was 32 years old and carries the TTN mutation, but without clinical symptoms of cardiomyopathy. B-mode ultrasonography showed a slight increase in left ventricular diameter (LVEF 67%, FS 37%, left ventricular diastolic diameter 56 mm). The normal left ventricular diastolic diameter scale is 35−55 mm in female. Individual III-4 was 30 years old and had received a diagnosis of DCM 4 years previously. Ultrasonography demonstrated a very large left ventricular diastolic diameter of about 72 mm with left ventricular dysfunction (LVEF 37%, FS 18%). Cardiac MRI showed a thin left ventricular wall (septum 6 mm, lateral wall 4 mm) and left ventricular noncompaction. Late gadolinium enhancement MRI showed that there were obvious fibrosis signs in the left ventricular wall and septum in individuals II-2 and III-4 (see Figure 3), which may be associated with persistent arterial fibrillation or FPVC. Individual II-4 was 59 years old and he had received a diagnosis of DCM 4 years previously. He declined to participate in genome sequencing. Individual IV-1 is a carrier of this TTN mutation, but he was 10 years old, that’s too early to predict his symptoms. Individuals I-1 and I-2 both died of esophageal cancer at the ages of 70 and 66 years, respectively, more than 15 years previously. Anecdotally, individual I-1 had gasped heavily for several years before he died, and it is uncertain whether he had DCM because of limited clinical data.

Discussion
We screened three DCM pedigrees in the province of Henan, China. As far as know, there have been no other reports of the DCM-causing TTN mutations in this province in China. Although we studied only three pedigrees, fewer than 50 persons, many other similar TTN mutations are constantly being discovered. This demonstrates that TTN mutation is also the main cause of DCM in Henan province, and we may have ignored this important mutation factor before. Therefore, we need more investigations to find and target those carriers of a TTN mutation before their symptoms get worse.
Cardiomyopathy is often thought to be an autosomal dominant inherited disease with unknown penetrance. DCM has a prevalence of 1 in 2500 individuals, while HCM occurs in less than 1 in 500 individuals [10, 11]. DCM and HCM are the most commonly inherited cardiomyopathies in the general population. Additionally, some reports have suggested that the prevalence of DCM is underestimated and may be higher than that of HCM [12]. Most of the genes associated with these diseases, such as MYBPC3, MYH7, MYH6, TNNI3, ACTC1, TNNC1, TNNT2, and TTN, encode sarcomere proteins [13].
Among all the familial DCM patients, TTN-truncating mutations are the most common [14]: 18% of nonfamilial and 25% of familial DCM patients carry truncating mutations in TTN [15]. From the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar), we know that most of the pathogenic TTN mutations result from frameshift and nonsense mutations. In families A and B, nonsense mutations result in the normal amino acid changing to termination codons. In family C, a splice site mutation happens near exon 215, disrupting RNA splicing, and as a result disrupting the coding sequence splicing. Consequently, an abnormal shorter messenger RNA (mRNA) is formed.
Nonsense-mediated mRNA decay is an mRNA surveillance system that eliminates abnormal mRNAs, especially those with premature termination codons [16, 17]. It can ideally eliminate the nonfunctional TTN mRNA in the three families, resulting in haploinsufficiency. This will subsequently disturb the cooperation of the sarcomere proteins and lead to cardiac dysfunction. So TTN truncation (including nonsense and splice site mutations) can be highly pathogenic, and the TTN mutations in these families are likely the cause of their cardiac symptoms.
To further confirm the pathogenicity of these mutations, we assessed the mutation and clinical data in every family using co-segregation analysis and used the criteria of the American College of Medical Genetics and Genomics–Association for Molecular Pathology guidelines [18]. We concluded that all three mutations we identified are classed as pathogenic very strong (PSV1), and are likely pathogenic. In the ClinVar database, none of the three mutations were reported before. Therefore, this is the first time these three mutations have been reported. What is more, this is the first report of TTN mutation resulting in DCM in Henan province in China.
It was demonstrated that individuals with a TTN truncation have a higher risk of DCM, but also persistent arterial fibrillation and ventricular tachycardia, and consequently require the assistance of a cardiac resynchronization therapy defibrillator (CRT-D) or an implantable cardioverter-defibrillator (ICD) [19]. In our study, two families (A and C) had persistent arterial fibrillation or ventricular tachycardia (Figure 4), but neither of the families used an ICD or a CRT-D. A previous report indicated that of all the causes of death in cardiomyopathy patients, cancer is a second element [20]. Meanwhile, more and more reports demonstrate that the tumor suppressor p53 gene (TP53) plays an important role in the development of cardiomyopathy [21, 22]. In family C, individuals I-1 and I-2 both died of esophageal cancer, and although we have no idea of their genotype, their offsprings harbor TTN mutations. They are probably inherited from them. Notably, individual I-1 had experienced shortness of breath for many years before he died. Unfortunately, their clinical data and their gene sequences were unavailable, so it is impossible to confirm their diagnoses.
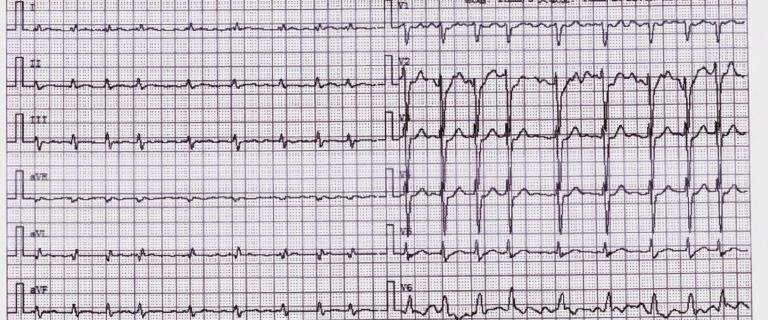
Electrocardiogram Results for Individual II-2 from Family C.
Persistent atrial fibrillation was detected by electrocardiogram.
In conclusion, we have identified three new likely pathogenic TTN mutations—c.G20137T,c. G52522T,c.44610-2A>C in exons 154, 79, and 215, respectively—in three families, and have broadened the TTN mutation spectrum in cardiomyopathy patients. It has been demonstrated that gene mutations play a very important role in DCM [23]. As all the third-generation individuals are young in the three pedigrees, it is of great importance to conduct long-term follow-up studies with them and to take action immediately if symptoms of DCM develop. Recently, for the first time to our knowledge, we successfully prevented the genetic transmission of a cardiomyopathy mutation from a serious case of HCM in a female patient through preimplantation genetic diagnosis (data not shown). We expect that this process will not be limited to HCM, and that more and more families with inherited diseases will benefit from it.