
Introduction
Marfan syndrome (MFS; OMIM 154700) is a relatively common autosomal dominantly inherited disease and multiple systemic disease that causes mainly skeletal, cardiovascular, and ocular defects with a prevalence of 1 in 3000 to 1/ in 5000 [1–3]. The typical symptoms include ectopia lentis, myopia, aortic dilatation or dissection, wrist and/or thumb sign, and hindfoot deformity [3]. The most common reason why untreated MFS patients die is cardiovascular complications, such as aortic dissection and rupture. The FBN1 gene has been identified as the major causative gene, which accounts for more than 90% of MFS patients [3, 4].
FBN1 is a large gene consisting of 65 exons and encodes a 2871-amino acid structural protein, fibrillin 1, which contains 47 epidermal growth factor (EGF)-like domains and nine transforming growth factor β–binding protein–like (TB) domains [5–7]. Fibrillin 1 is a major component of connective tissue in the body. There are more than 3000 mutations in the FBN1 gene that have been identified to be related to MFS, most of which are missense mutations [8]. What is more, there is a large variation in the clinical features of patients with MFS. Therefore, more research is needed to enrich the phenotypes and genotypes of MFS.
Here we report three mutations in three Chinese families with MFS found by genome sequencing and present their clinical features.
Materials and Methods
Patients and Clinical Information
This study was approved by the Medical Ethics Committee of the First Affiliated Hospital of Zhengzhou University. All patients in this study were recruited for our research, and written consent was obtained from the patients and/or their family members. Clinical diagnosis was established according to the revised Ghent nosology by physicians [3]. Physical examination of patients was performed, including cardiac ultrasonography, electrocardiogram, and computed tomography or magnetic resonance imaging for the cardiovascular system, radiology for the skeletal system, and slit-lamp examination for the ocular system. The clinical information was reviewed but complete data were not available for all family members.
Genomic DNA Preparation
Peripheral blood samples were collected for blood cells and were then used for extraction of genomic DNA from patients and their family members with a QIAamp DNA mini kit (Qiagen) according to the manufacturer’s protocol. Whole exomes of each proband were enriched with a SureSelect Human All Exon V5 kit (Agilent, Santa Clara, CA, USA), and the sequencing was conducted with an Illumina HiSeq 2500 sequencing system (Zheng Zhou Apply Medical Laboratory).
Next-Generation Sequencing and Analysis
All the reads were qualified by FastQC (version 0.11.4) with default parameters. The remaining reads were mapped to the hg19 human reference genome with BWA (version 0.7.9a-r786) and processed with Picard (version 2.20.0) to mark duplicate reads. Realignment around indels and recalibration of quality scores and variant calling were conducted according to GATK best practice recommendations (https://software.broadinstitute.org/gatk/best-practices/). Each variant was removed if it cannot fit following criteria: with more than 5 reads supported, the MAF of it was ≥5% and variant quality score ≥20. Also, common nondisease known genetic variants from genome variation project data (1000 Genomes Project, Exome Aggregation Consortium available at ftp://ftp.broadinstitute.org/pub/ExAC_release/current/ExAC.r0.3.1.sites.vep.vcf.gz. DiscovEHR (http://www.discovehrshare.com), and the Genome Aggregation Database (http://gnomad.broadinstitute.org) and variants with minor allele frequency of 0.01 or greater were excluded. In the final stage of analysis, variants were annotated with use of different databases, such as dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP), dbscSNV (http://asia.ensembl.org/info/docs/tools/vep/script/vep_plugins.html#dbscsnv), and OMIM (https://www.omim.org). The interpretation of sequence variants was according to American College of Medical Genetics and Genomics (ACMG) standards and guidelines.
Sanger Sequencing
For each mutation, a pair of primers were designed with Primer3. The genomic DNA was amplified as follows: 95 °C for 5 min; 30 cycles of 95 °C for 30 s and 60 °C for 1 min; 72 °C for 25 min. The amplicons were sent for Sanger sequencing. The primers are as follows: primer 1, forward CCTGGTGTAAAGGTGACTCCC, reverse CTAGCCCATCATCCCGAGTG; primer 2, forward AACTTACTTCAGACGGGCAGAG, reverse GGCCCAGAACTCCTCCAAATA; primer 3, forward CCTCTGGTTTCTGGGCTTGT, reverse TGAGAATCCAGCACAGGCAA.
Functional Prediction of the Mutations
PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2) was used to predict the potential effect of a specific mutation. The clinical effects of mutations were searched for in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
Results
Major Clinical Characteristics in the Families
The pedigrees of the families are shown in Figure 1, and the clinical information is summarized in Table 1. All probands displayed cardiovascular system abnormalities, including abdominal aortic aneurysm. Proband 1 had a phenotype of aortic root dilation and aortic dissection but no other abnormalities and family history of MFS. However, there were no typical symptoms in his sister (III:3) and his son (IV:4). In proband 2, MFS with abdominal aortic aneurysm and wrist and thumb sign was diagnosed at the age of 38 years, but no similar abnormalities have been observed in her two daughters (III:5, III:6) and her older brother (II:2), except that one daughter (III:5) had severe myopia and her older brother (II:2) had increased arm/height. Proband 3 had classic abdominal aortic aneurysm and severe myopia rather than others in her family.
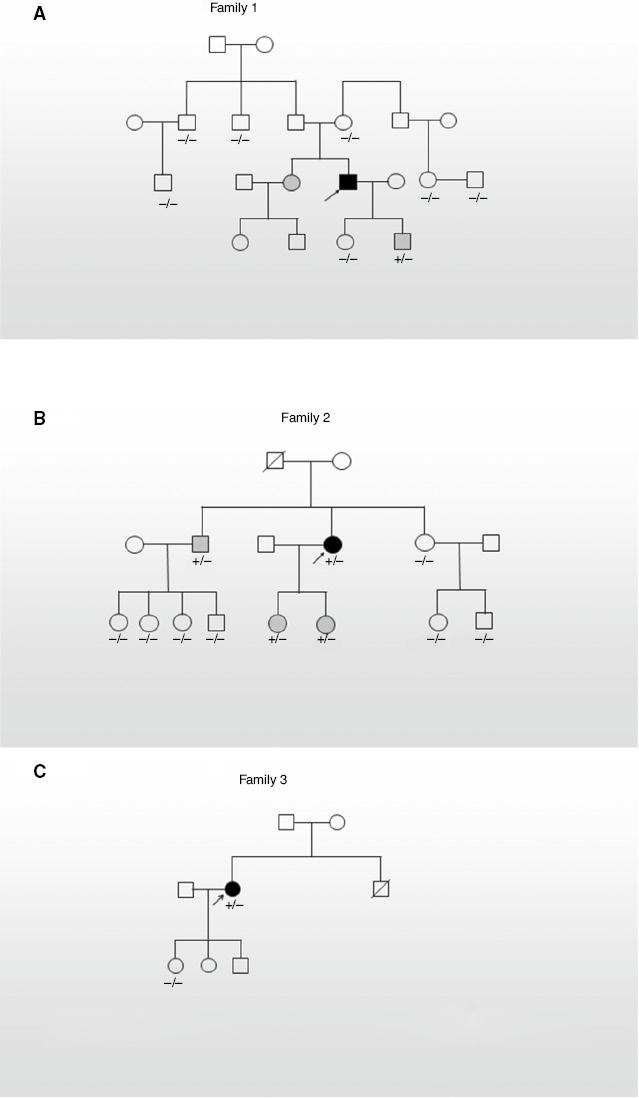
(A–C) Indicate the pedigrees for family1, family 2 and family 3, respectively.
Squares indicate males and circles indicates females. Dark symbols mean patients. Gray symbols mean a heterozygous mutation but no typical symptoms. +/− and −/− mean heterozygous mutation and wild-type genotype, respectively. The molecular information on the members without a marked genotype is not available.
Clinical Information on the Families.
| Family | Patient | Sex | Age (years) | Height (cm) | Cardiovascular involvement | Ocular involvement | Skeletal involvement |
|---|---|---|---|---|---|---|---|
| 1 | III:3 | F | 50 | 172 | Moderate MVR | None | None |
| III:4 (proband 1) | M | 48 | 178 | Aortic dilatation, aortic dissection | None | None | |
| IV:4 | M | 22 | 184 | None | None | None | |
| 2 | II:2 | M | 45 | 180 | None | None | IAH |
| II:4 (proband 2) | F | 38 | 181 | AAA | None | Wrist and thumb sign | |
| III:5 | F | 16 | 180 | None | Severe myopia | None | |
| III:6 | F | 9 | 140 | None | None | None | |
| 3 | II:2 (proband 3) | F | 41 | 180 | AAA | Severe myopia | None |
AAA, abdominal aortic aneurysm; F, female; IAH, increased arm/height; M, male; MVR, mitral valve regurgitation.
Mutation Analysis
We performed whole-exome sequencing of three families and identified three missense mutations in the FBN1 gene, including one novel mutation (2125G > A) and two known mutations (Table 2). All these mutations were validated in probands and their affected family members by Sanger sequencing, except for 6325C > T (Figure 2). The novel mutation 2125G > A (Ala709Thr) in proband 1 was located in the region coding for TB domain and was found in his sister and his son. Although this novel variant was defined as of uncertain significance according to the ACMG, it is predicted to be probably damaging by PolyPhen-2. The previously reported mutation 4786C > T (Arg1596ter) identified in proband 2 resulted in a premature termination codon, which led to deletion of a peptide fragment of about 1000 amino acids, which is usually considered to be pathogenic [9–13]. This mutation was also identified in her brother and her two children, but not in her sister. The other previously known mutation, 6325C > T (Gln2109ter), detected in proband 3 has also been reported in several other MFS patients, and is defined as a pathogenic mutation according to ACMG guidelines [14, 15].

Mutation Analysis.
| Proband | Gene | Nucleotide mutation | Type | Exon | Domain | Amino acid alteration | New variant | Effect | PolyPhen-2 | ClinVar | ACMG |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | FBN1 | c.2125G > A | Missense | 18 | TB domain 3 | p.Ala709Thr | Yes | Substitution | Probably damaging | NA | Uncertain significance |
| 2 | FBN1 | c.4786C > T | Nonsense | 39 | p.Arg1596ter | No | PTC | NA | Pathogenic | Likely pathogenic | |
| 3 | FBN1 | c.6325C > T | Nonsense | 52 | TB domain 8 | p.Gln2109ter | No | PTC | NA | Pathogenic | Pathogenic |
ACMG, American College of Medical Genetics and Genomics; NA, not available; PTC, premature termination codon, TB, transforming growth factor β–binding protein–like.
Discussion
MFS is a complex connective tissue disease inherited in an autosomal dominant fashion and primarily involves the ocular, skeletal, and cardiovascular systems. It usually result in decreased life expectancy mainly because of the cardiovascular complications, including aortic root aneurysm and aortic dissection [3, 16]. What is more, there are a wide variety of clinical features in MFS among different mutations of genes and their types and location [17–19]. In this study, all probands did not show all the typical symptoms of MFS, and the symptoms of mutation carriers among their relatives were even less obvious, especially in one relative who was 40 years old. However, the genotyping data showed that these relatives potentially have MFS. This is because two of the three mutations we found are known pathogenic mutation sites, which may lead to the complete loss of the function of the gene, and the novel mutation was predicted to cause damage to the function of FBN1. Heterozygous mutations do not account for this phenomenon, because cases of heterozygous mutations have been reported in typical patients. All these results indicated that MFS has a more complex pathogenic mechanism. This phenomenon could also lead to misdiagnosis and missed diagnosis of MFS, and thus underestimation of the frequency of MFS in the population. Therefore, genetic testing should be more frequently used in the clinical detection of suspected MFS. The collection/analysis of new mutation sites and MFS cases will be helpful for the analysis of pathogenesis, and will also provide more references for the diagnosis and treatment of this disease, which is one of the important aspects of this study.
FBN1 is the gene that is most commonly mutated in MFS. It encodes a structural protein, fibrillin 1, that contains 47 EGF-like domains and nine TB domains consisting of conserved cysteine residues [20]. The EGF domains bind calcium and mediate the interaction of fibrillin 1 and ligands that is important for microfibril assembly, and TB domains are associated with interchain disulfide-bridge formation between fibrillin 1 molecules [21]. Most FBN1 mutations occur in exons, especially in the region coding for EGF domains [22]. Substitution of cysteine residues is critical in the pathogenesis of MFS, and it seems there is a strong correlation between ectopia lentis and pathogenic variants in the cysteine residues [4, 17, 23–25]. It was indicated that mutations in exons 1–10 were associated with no cardiovascular complications, but mutations in exons 24–32, encoding TB domain 3 and eight calcium-binding EGF domains, usually caused severe cardiac manifestations [26–28]. Many studies showed a tendency for missense mutations at the 5′ end of the FBN1 gene to lead to ectopia lentis and for nonsense mutations at the 3′ end of the FBN1 gene to lead to early-onset aortic aneurysms/dissection and sudden death [17, 27]. It was reported that the prevalence of major cardiovascular involvements was higher in MFS patients with FBN1 mutations in exons 43–65 [29].
In this report, three FBN1 mutations were identified in three Chinese families by genome sequencing, including one novel missense mutation. Proband 1 carries a novel mutation, 2125G > A, that results in substitution of a threonine residue for alanine and has a phenotype of aortic root dilation and aortic dissection. It was interesting that no similar MFS-like symptoms could be found in his sister (III:3) and his son (IV:4), who also carried the same mutation. The nonsense mutation (4786C > T) found in proband 2, leading to a premature termination codon, has been reported previously to cause similar phenotypes of cardiovascular and skeletal abnormalities, including abdominal aortic aneurysm and wrist and thumb sign [9–13]. Her brother (II:2) and two daughters carried the same mutation, but no typical abnormalities of MFS could be observed, except that her brother was thin and tall and had relatively big feet and hands, and her older daughter had severe myopia. Finally, proband 3 with the previously reported variant 6325C > T exhibited apparent clinical features including abdominal aortic aneurysm and severe myopia although her parents were healthy [14, 15]. Even though there was no clinical and molecular information onher brother (II:3), he may have carried the same mutation because of his sudden death when he played basketball years ago. In summary, we found some carriers of known disease-causing mutations and a novel mutation with only slightly abnormal appearance, such as thin and tall, and myopia, but no typical MFS features could be observed. There is an apparent characteristic that most of them are young, which suggests that age plays an important role in the onset of MFS, and it is possible that they may exhibit MFS in the future. Furthermore, there were no typical symptoms of MFS in member II:2 in family 2, even though he is 45 years old. All findings indicated that some other factors are required for the onset of MFS, and more attention should be paid to the investigation of MFS cases to clarify the whole mechanism. Besides, it is of great importance to apply new methods, especially genome sequencing, to identify potential MFS patients so that they can receive proper medical treatment and care in a timely manner.
All in all, our research provides more information on the FBN1 mutation spectrum and enriches the phenotype–genotype correlation between identified mutations of FBN1 and MFS.