
1. INTRODUCTION
Podophyllotoxin (PPT), which was first isolated from the roots and rhizomes of Podophyllum species in 1880, is an aryltetralin-type lignan with anticancer, antineoplastic, antioxidant, antibacterial, cytotoxic, and immunosuppressive properties [1–7]. PPT extract has been reported to be a remedy for various medical conditions, such as gonorrhea, tuberculosis, menstrual disorders, psoriasis, dropsy, cough, syphilis, and venereal warts [8–12]. Despite being recommended as a first-line drug for the treatment of condyloma acuminatum by the World Health Organization in 1990, PPT cannot be administered orally or intravenously due to its severe systemic in vivo toxicity. Indeed, PPT is only involved in current clinical settings as a Chinese herbal curative ingredient or a precursor that is chemically modified into antitumor drugs [13, 14].
The chemical structure of PPT was established in the 1950s and revealed four consecutive chiral centers and four virtually planar fused rings [15]. The configuration at the C-4 position on PPT is critical for antitumor activity because derivatives with an α-configuration are potential tubulin polymerization inhibitors, whereas derivatives with a β-configuration are possible topoisomerase II inhibitors ( Figure 1 ) [16–18].

The mechanisms underlying PPT antitumor activity are associated with multiple signaling pathways, including the c-Jun N-terminal kinase (JNK)/p38 MAPK and phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathways ( Figure 2 ). Apart from causing DNA damage by inhibiting tubulin polymerization, PPT may also inhibit tumor cell growth by activating apoptosis-related molecules and the endoplasmic reticulum stress signaling pathway, most likely via interactions with cyclin B, p53 protein, JNK, and PI3K ( Figure 3 ) [19–23].

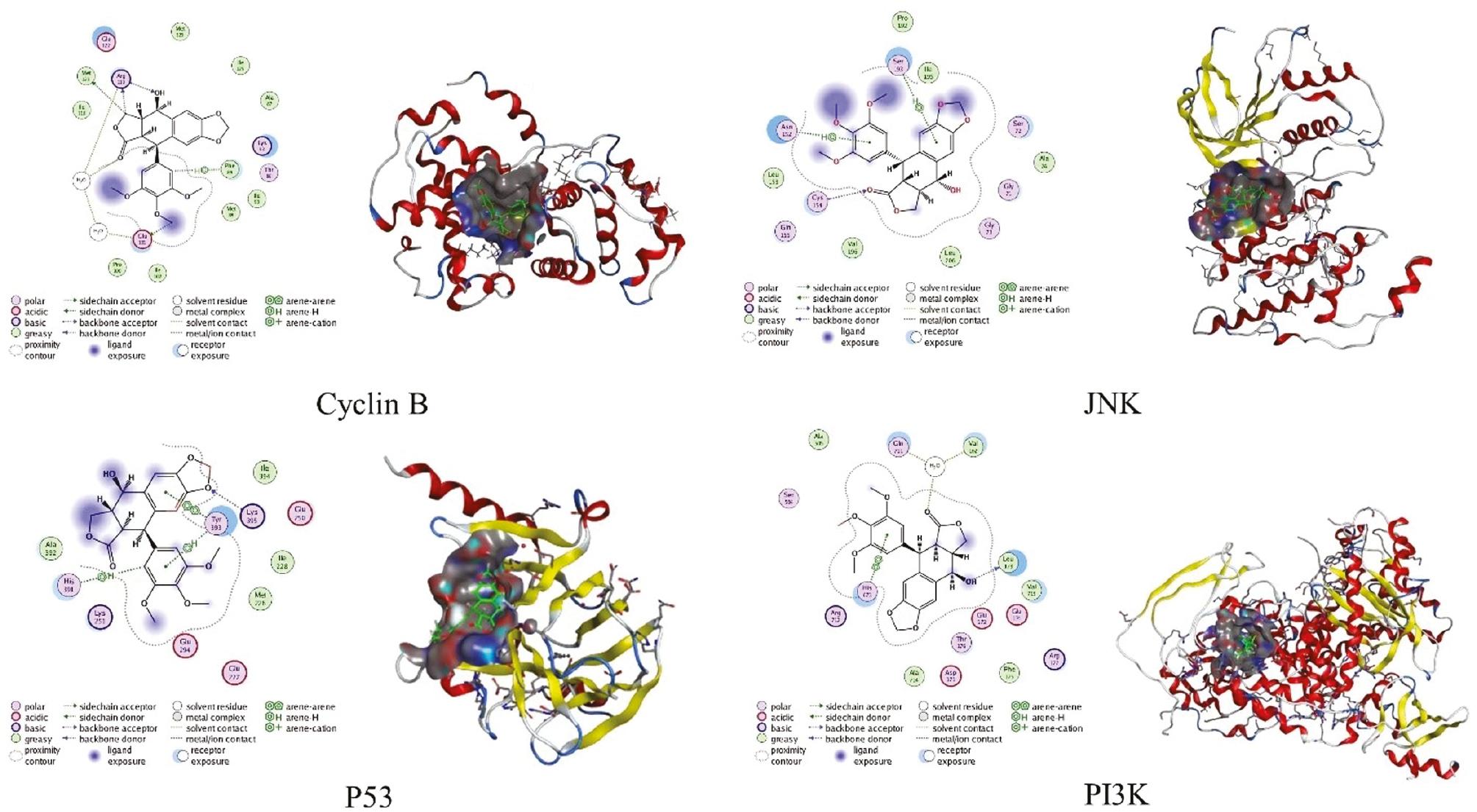
Three-dimensional structures of primarily targeted proteins and theorized two-dimensional schematic representations of the proteins and PPT complex interactions.
Unfortunately, use of almost all PPT derivatives, including FDA-approved drugs (etoposide and teniposide), are severely limited in the clinical setting due to poor solubility, systemic toxicity, drug resistance, and low bioavailability [24–26].
With the advent of nanotechnology, nano-based drug delivery systems (10–1000 nm) ranging from inorganic and carbon-based nanomaterials to lipid-based and polymeric nanomaterials have emerged as the mainstream nanotechnology application in medicine [27, 28]. Compared to conventional drug administration approaches, nano-based drug delivery helps to improve drug bioavailability, increase drug stability, and achieve controlled release, all of which improve the drug therapeutic index, especially demonstrating unique benefits for water-insoluble drugs, thus providing a new approach to insoluble drug administration [29–31].
Macromolecules are generally cleared by the lymphatic system in healthy tissues. In solid tissues, however, the rapid proliferation of endothelial cells and decreased number of pericytes result in large pores in the tumor vasculature, which leads to high vascular permeability and allows macromolecules, such as nanoparticles, to pass into tumors. Furthermore, the impaired lymphatic system in solid tumors cannot clear nanoparticles, which traps nanoparticles in tumors at higher concentrations than in plasma or other tissues; this phenomenon is known as the enhanced permeability and retention (EPR) effect [32]. Nanocarriers can achieve passive tumor targeting through the EPR effect and be modified with specific ligands for active targeting, which improves the biodistribution of conventional chemotherapeutic agents and facilitates aggregation and cell uptake in lesion tissues, thereby reducing drug toxicity in healthy cells [33]. Moreover, as the use of combined pharmacotherapy increases, rationally designed nano-preparations not only acquire synergistic delivery of PPT with other drugs or small molecules, but also improves the targeting effect, resulting in enhanced efficacy and reduced toxicity.
Few reviews on advances in PPT nanodrug formulations have been published. Considering the fascinating aspects of PPT in pharmacology and preclinical medicine, we would like to present a survey to gain a comprehensive understanding of the progress in nanotechnology for PPT delivery to provide more reliable use in the clinical setting and to shed light on further PPT formulation research.
2. PODOPHYLLOTOXIN-LOADED NANOCARRIERS
Nanomaterials for drug delivery are categorized into two types based on structure and composition: organic sources (lipid- and polymer-based nanomaterials); and inorganic sources (metallic and non-metallic nanocarriers). In the present review we have focused on nanocarriers for PPT delivery (predominantly lipid- and polymer-based nanocarriers), as well as inorganic nanocarriers and cyclodextrin inclusions.
2.1 Lipid-based nanocarriers
Lipid-based nanoparticles (NPs) are the most-studied nanocarriers and include liposomes, solid lipid NPs, and nanostructured lipid carriers. Lipid-based NPs have the advantage of being the least toxic for in vivo use and are therefore one of the promising candidates for drug delivery [34].
2.1.1 Liposomes
Liposomes are spherical vesicles with a large aqueous core and a biocompatible lipid shell composed of a single or multiple natural or manufactured lipid bilayers (10–1000 nm) [35–39]. Liposomes have become one of the most applicable nanoscale drug delivery vehicles for the following reasons: non-toxicity; non-immunogenicity; excellent selectivity; continuous drug release with prolonged duration; and ease of degradation in vivo. Pharmaceutical researchers have shown an enormous interest in liposome applications involving the delivery of PPT and PPT derivatives [40, 41].
PPT-dipalmitoylphosphatidylcholine (PPT-DPPC) and PPT-soya lecithin liposomes were constructed with embedding ratios of 73.8% and 79.1%, respectively, using the reverse phase evaporation technique [42]. To further optimize the preparation conditions of PPT liposomes (PPT-Lips) and examine the effects on PC3 cells, PPT-Lips were created using the thin-film dispersion method with varying component ratios. The optimum preparation conditions optimized by the response surface methodology (RSM) were a cholesterol-to-lecithin ratio of 3.6:40 (w/w), a lipid-to-drug ratio of 15.8:1 (w/w), and an ultrasonic intensity of 35% (total power = 400 W). The experimental encapsulation efficiency (EE) of PPT-Lips was up to 90.425%, with an average well-dispersible spherical particle size of 106 nm and a zeta potential of −10.1 mV. A gradual and time-dependent pattern of PPT from liposomes was found in in vitro drug release, with a cumulative release amount of up to 70.3% in 24 h. Cell viability tests on PC3 cells revealed that PPT-Lips exhibited more effective anticancer activity than free PPT [43].
Thin film technology was used to create a new amphiphilic prodrug dual PPT succinate glycerophosphocholine (Di-PPT-GPC)-assembled liposome with an average diameter of 160 nm [44]. Di-PPT-GPC liposomes significantly release PPT in a mildly acidic environment but have good stability under biological conditions according to in vitro release experiments. Data from the methyl tetrazolium (MTT) assay revealed that the liposomes are cytotoxic to MCF-7, HeLa, and U87 cells as free PPT. More importantly, an in vivo antitumor evaluation indicated that Di-PPT-GPC liposomes exhibit favorable tumor growth inhibition without side effects, which may pave the way for enhancing PPT therapeutic benefits in cancer therapy.
A dimeric-PPT phospholipid was then produced and self-assembled into Di-PPT-GPC liposomes, which display a cell membrane-like transport pattern with increased cellular uptake, excellent antitumor effectiveness, and minimal side effects [45].
Introducing peptide groups into lipids can prolong the in vivo half-life and enhance targeting ability. Peptide-based cationic lipids have shown greater superiority over other cationic lipids and therefore have potential application in improving the delivery of gene therapeutics [46–48]. To maximize the therapeutic effects of PPT and minimize systemic toxicity, PPT was conjugated with 4-(4-methyl-1-piperazinylmethyl) benzoic acid (PBA) via an enzymatically degradable amido linkage to form an amphiphilic PPT-PBA conjugate. The PPT-PBA conjugate and a tripeptide cationic lipid (CDO) were then self-assembled into drug-loaded lipid bilayer NPs (designated PPCNs) using a dried film-ultrasonic procedure [49]. In addition to improved targeted efficacy and fewer side effects than plain PPT, PPCNs also induce tumor cell apoptosis by downregulating the expression of programmed cell death ligand 1 (PD-L1) on tumor cells, thus displaying both immunologic and anticancer effects ( Figure 4 ).
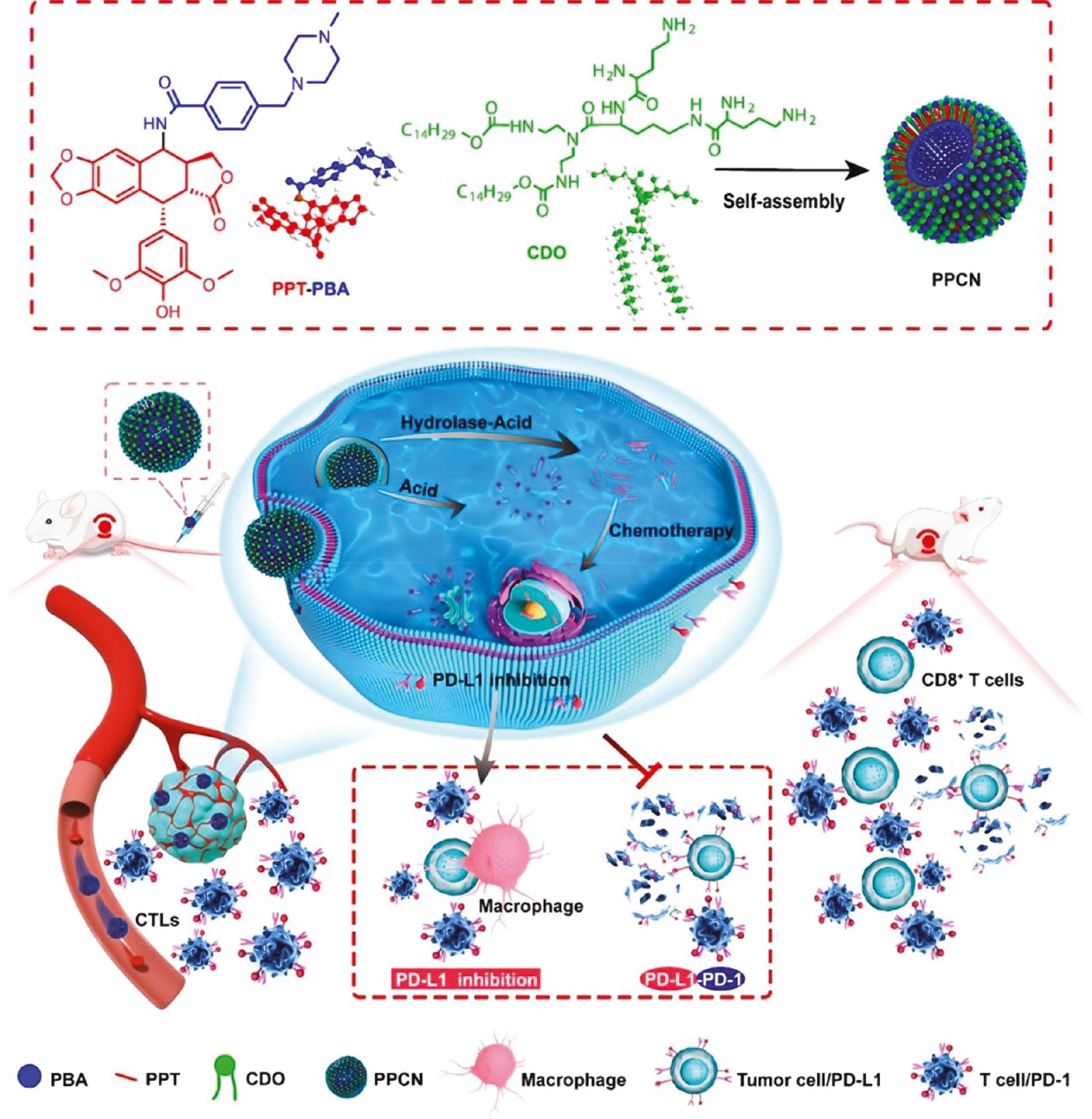
PPCNs were generated by self-assembly of CDO and a PPT−PBA conjugate synthesized from PPT and PBA.
The drug-loaded nanoparticles were injected into tumor-bearing mice via the tail vein. The PPCNs not only released PPT for chemotherapy but also prevented tumor cell immune escape by inhibiting PD-L1 production, thereby improving the sensitivity of tumor cells to cytotoxic T lymphocytes (adapted from ref. [49]).
New generation ultra-deformable liposomes, also known as transfersomes, are composed of phospholipids and edge activators. Transfersomes have emerged as a promising transdermal delivery system due to ultra-flexibility, which enables the transfersome to cross the stratum corneum barrier and penetrate orifices that are 5-10 times smaller than the transfersome diameter [50]. Recently, PPT-loaded L-cysteine-modified transfersomes were successfully fabricated via the thin membrane dispersion method for enhanced epidermal delivery, which reduced adverse effects caused by systemic absorption. Due to retention in the stratum corneum and epidermis, the PPT-loaded L-cysteine-modified transfersome is a novel drug delivery system for the topical treatment of genital warts [51].
Liposomes, the most successful nanoscale drug delivery carriers utilized in clinical applications, might encapsulate PPT to boost its aqueous solubility and alleviate toxic side effects, along with enhancing anti-tumor effects. These liposomes may also lower the in vivo clearance of PPT, prolong the retention time, and increase the in vivo and in vitro stability.
2.1.2 Solid lipid nanoparticles (SLNs)
SLNs are colloidal dispersions of non-polar lipids with diameters ranging from 50–1000 nm. SLNs are solid at ambient and body temperatures, have several intriguing properties, such as excellent biocompatibility, biodegradability, and an easy preparation process, thus making SLNs well-positioned for mass production [52]. To address concerns, such as drug leakage and burst release, SLNs were created as an alternative to conventional colloidal drug carriers. The application of solid lipids significantly decreases the mobility of incorporated drugs into the lipid matrix, which prevents particle coalescence, improves stability, limits drug protuberance into the emulsifier film, and results in sustained drug release [53–56].
To evaluate SLNs as the topical carrier for epidermal targeting of PPT, a PPT-loaded SLN (P-SLN) with high-pressure homogenization was prepared using poloxamer 188 as a surfactant [57]. To further evaluate the influence of surfactants and other factors on SLN skin targeting, another PPT-loaded SLN (T-SLN) was prepared using polysorbate 80 rather than poloxamer 188 as surfactant. An in vitro permeation study showed that P-SLNs increased the cumulative amount of PPT in porcine skin 3.48-fold compared to 0.15% PPT tincture. T-SLNs did not exhibit a high cumulative amount of PPT compared to P-SLNs, although P-SLNs and T-SLNs did not have systemic uptake of PPT. Penetration of P-SLNs with low particle size (average = 73.4 nm) into the stratum corneum along the skin surface furrow and the controlled release of PPT might lead to epidermal targeting. P-SLNs provide a good epidermal targeting effect and may be a promising carrier for topical delivery of PPT.
Novel PPT-loaded solid lipid nanoparticles (PPT-SLNs) with an approximate particle size of 50 nm made from stearic acid, lecithin, and myrj59 were prepared using a solvent emulsification-evaporation method and the anti-tumor function was determined [58]. PPT was released from the SLNs in a slow but time-dependent manner in an in vitro drug release study. PPT-SLNs exhibited more anti-tumor potency against 293T and HeLa cells compared to unconjugated PPT. In addition, PPT-SLNs enhanced the apoptotic and cellular uptake processes on tumor cells with less toxicity to healthy cells.
As an alternative to conventional carriers for chemotherapeutic agents and cosmetic active ingredients, SLNs protect labile compounds from chemical degradation, maintain moisture in the skin, favor drug penetration into the skin, provide controlled drug release over a prolonged period, and reduce systemic absorption [59–62]. However, the low drug loading efficiency and elevated risk of expulsion of the drug from SLN formulations due to polymorphic transitions during storage limit applicability.
2.1.3 Nanostructured lipid carriers (NLCs)
NLCs, which use mixes of solid and liquid lipids to generate an amorphous solid matrix at body and room temperatures, are the next generation of lipid NPs. NLCs outperform SLNs with respect to stability, loading capacity, and drug expulsion during storage. In contrast to the perfect crystalline solid matrix formed in SLNs, which limit the amount of loaded drug and lead to expulsion due to limited spatial capacity, the incorporation of liquid lipids in the matrix considerably enhance the characteristics of the formulation, allowing for a greater load of the drug to be added [63–65].
The emulsion evaporation process was adopted to generate PPT-NLCs with a diameter of 85.6 ± 10.25 nm, which were subsequently tested for in vitro proliferation suppression on HPV-infected H8 cells. PPT-NLCs inhibit cell growth far more effectively than free PPT in a concentration- and time-dependent manner [66]. A corollary study showed that PPT-NLCs induce apoptosis in VK2/E6E7 cells, possibly by triggering the endoplasmic reticulum stress response, and upregulate GRP78, GRP94, and calpain2 mRNA and protein expression in a concentration-dependent manner [67].
Two PPT-NLCs were prepared by hot high-pressure homogenization using Compritol® 888 ATO, Labrasol®, Cremophor® RH40, and soybean phosphatidylcholine as nanostructured lipid carriers [68]. The particle diameter, encapsulation efficiency (EE), and zeta potential differ as a function of the formulation composition ratio. Both PPT-NLCs were safe for topical application and exhibited high in vitro and in vivo rat skin deposit amounts, while the PPT-NLC with a smaller mean size had better skin targeting ability. The EE with a bigger particle size reached 93.5%; however, the PPT content in both PPT-NLCs were relatively low (0.33% and 0.49%, respectively).
To assess the physicochemical quality and efficacy in the treatment of condyloma acuminatum (CA), a new PPT-NLC was developed via emulsion evaporation and a low-temperature solidification approach [69]. When compared to PPT tincture, the aforementioned PPT-NLCs with a diameter of approximately 178.5 nm demonstrated favorable physicochemical characteristics for transmucosal delivery, thus providing sustained release and controlled delivery in the cervical mucosal tissue, inhibiting the growth of VK2/E6E7 cells via G2/M arrest, suppressing cell proliferation more efficiently, and causing less inflammatory cytokine production in the cervical mucous. As a result, NLCs were confirmed to decrease the systemic toxicity of PPT and enhance skin targeting, which are encouraging findings for applicability as a substitute carrier for PPT.
Liposomes, as classical examples of lipid-based nanoparticles primarily consisting of phospholipids, have been widely investigated for delivering drugs. However, concerns about the preparation process, including batch-to-batch variability, the necessity of sterilization, the desirability of increased encapsulation rates, particle size control, shelf life, and organic solvent residues, have severely hampered the development and application of liposomes. SLNs with a solid lipid core that reduces drug mobility and improves the controlled release of loaded drugs can be prepared at a lower cost and easily scaled up, although undesirable particle growth by agglomeration or coagulation may result in rapid “burst release” or expulsion of the drug. Instead of perfect lipid matrices as in SLNs, an imperfect crystal or amorphous structure is formed from solid and liquid lipids in NLCs that facilitates drug loading in the molecular form and clustered aggregates at lattice imperfections. Thus, lipid-based nanoparticles have been shown to be ideal carriers for targeted delivery and controlled release of PPT.
2.2 Polymeric NPs
Polymeric NPs, encompassing polymeric micelles, polymer-drug conjugates (polymeric prodrugs), and dendrimers, stand out among various NP-based drug delivery systems as a flexible class of nanocarriers that facilitate controlled drug delivery to diseased tissues [70].
2.2.1 Polymeric micelles
Polymeric micelles (PMs) are amphiphilic co-polymers with a hydrophobic core that can load various types of insoluble drugs. The hydrophilic shell of PMs can prevent drugs from interacting with the mononuclear phagocytic system and prolong circulation time in the blood. The distinct core-shell structure of PMs enables the identification of appropriate micellar carriers based on pharmacologic characteristics [71–73]. Targeted delivery of anticancer medications is achievable by connecting specific antibodies, ligands, or sensitive response systems in the hydrophilic chain segment of the micelles. PMs accumulate in solid tumors to achieve therapeutic effects, overcome biological barriers, and decrease the toxic side effects of anticancer treatments. PMs therefore represent the most reassuring nanoparticle drug delivery technique that overcomes the problem of non-targeted traditional chemotherapeutic medicines [74–77].
PPT coupled to stearic acid-g-chitosan oligosaccharide micelles (CSO-SA) by dialysis achieved the highest CSO-SA/PPT drug EE (up to 70.2%) when the drug feeding ratio was 10% [78]. CSO-SA/PPT micelles demonstrated sustained release performance as well as increased cytotoxicity against MCF-7, A549, and Bel-7402 cell lines when compared to the free PPT formulation, which might be attributed to quicker PPT transport into tumor cells mediated by CSO-SA micelles.
To improve the efficacy of PPT, Yadav et al. [79] encapsulated PPT into poly-D, L-lactide nanoparticles (PLA NPs) using the solvent evaporation method. The particle size of PPT-loaded PLA NPs was 100 ± 17 nm with an encapsulation efficiency of only 17%. An in vitro release study showed an initial burst release followed by slow and sustained release, and PPT-loaded PLA NPs showed higher in vitro cytotoxicity to A549 and CHO-K1 cells compared to pure PPT.
A redox/pH double-sensitive and tumor active-targeting polymer (Pep-PEG-SS-PPT or PEG-SS-PPT) was subsequently created, in which PPT was covalently coupled with T7-peptide (Pep)-modified polyethylene glycol (PEG) or mPEG through a disulfide bond [80]. The mixed micelles (Pep-SS-NPs), made by mingling Pep-PEG-SS-PPT with PEG-SS-PPT, have good size uniformity and high stability in serum solution. After uptake by cancer cells, the prodrug micelles are enriched in the tumor via Pep-mediated active-tumor targeting and rapidly release PPT due to the disulfide bond disassociation in response to the high concentration of glutathione (GSH) and low pH. Pep-SS-NPs have an exceptional cell proliferation inhibition effect against two drug-resistance cell lines (MCF-7/ADR and A549/PTX) and increase the maximum tolerated dose 5.3-fold compared to free PPT.
For the efficient delivery of PPT to overcome multi-drug resistance (MDR), pH/redox cascade-sensitive multiscale nanoparticles with size- and charge-changeable properties were developed containing a charge-reversible polymer (PEG and dimethylmaleic acid-grafted polyallylamine (PEG-PAH-DMA)) shell and a redox-sensitive small-sized dendrimeric PPT-prodrug (PPT conjugated with polyamidoamine (PAMAM) dendrimers with a disulfide linkage (PPT-ss-PAMAM)) core [81]. This negative-charged polymeric prodrug reverts to a positive charge in a mildly acidic tumor environment (pH 6.5), which leads to the release of positive PAMAM-ss-PPT via electrostatic repulsion and completely releases PPT under elevated intracellular GSH conditions in tumors ( Figure 5 ). The nanomedicines effectively penetrate xenografted A549 paclitaxel-resistant lung cancer cells and inhibit tumor proliferation with negligible toxicity.
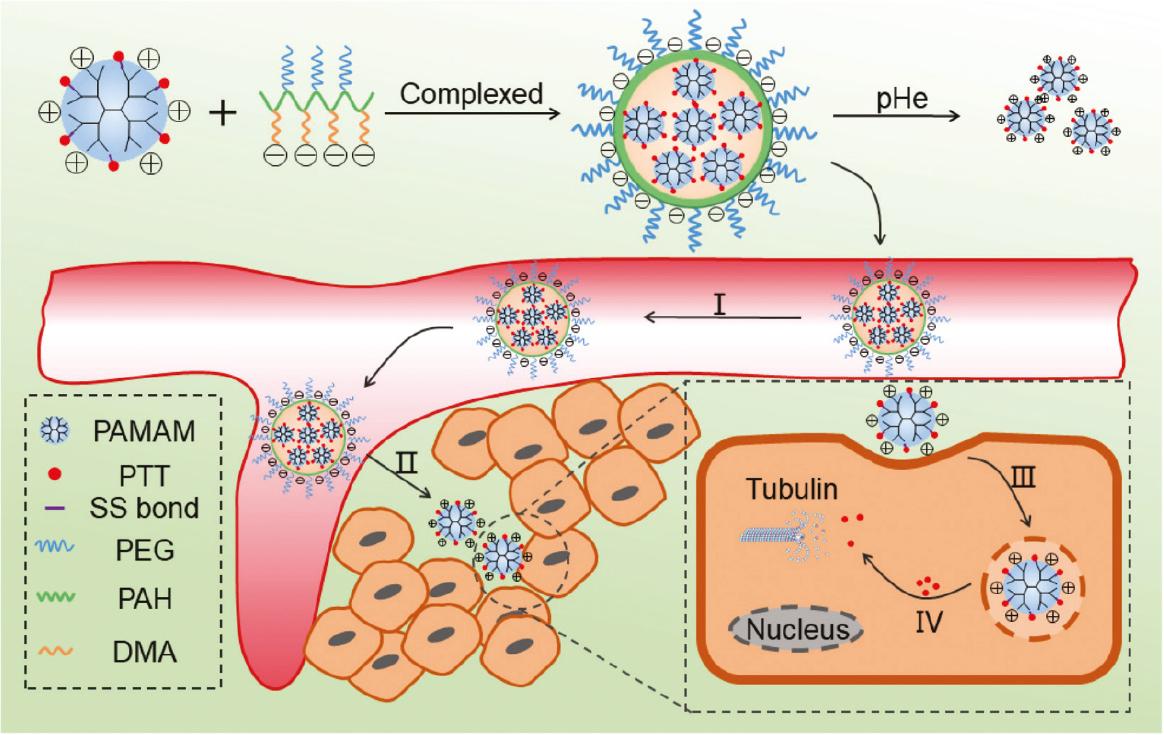
Preparation of DMA-NPs that effectively overcome physiologic barriers.
DMA-NPs complexed to a negatively-charged polymer (PEG and DMA-modified PAH [PEG-PAH-DMA]) and a positively-charged PPT prodrug (PAMAM-ss-PPT) through electrostatic interactions achieved a cascade drug delivery dependent on tumor microenvironment-triggered size and charge transformation. (I) Longer blood circulation times; (II) size shrinkage and charge reversal; (III) positive charge enhancing the cell uptake; (IV) GSH-triggered PPT release (adapted from ref. [81]).
A PPT polymeric prodrug micelle (PCDMA) with charge-conversion capability and self-acceleration drug release function was subsequently fabricated via self-assembly [82]. These PPT-loaded polymeric micelles, constructed by esterized PPT (TK-PPT) conjugated with mPEG-block-poly(L-lysine) [MPEG-b-PLL] were then modified by dimethylmaleic anhydride (DMA). After encapsulating cucurbitacin B (CuB), the aforementioned compound self-assembled into a PCDMA. The surface charge of PCDMA immediately reversed to positive in the tumor extracellular environment to facilitate cellular uptake. The PCDMA was subsequently degraded to release PPT and CuB in response to high level of intracellular reactive oxygen species (ROS). The released CuB generates ROS, which in turn accelerates the release of PPT and CuB. Eventually, the released PPT kills MDR cancer cells. In vitro and in vivo studies demonstrated that PCDMA not only effectively boost cell uptake via tumor acidity-triggering charge switching and ROS-activating drug release, but also efficiently replenishes intracellular ROS to achieve rapid and complete drug release. PCDMA significantly improves the maximum tolerated dose of free PPT, which substantially enhances tumor treatment efficacy with low in vivo toxicity. Hence, the pH/ROS dual-responsive PCDMA micelles with surface charge-reversal and self-amplifying ROS-response drug release provide an excellent platform for plausible MDR cancer treatment.
Recently, Li et al. [83, 84] used a one-step esterification reaction to construct a pH-sensitive PPT prodrug (HA-CO-O-PPT) by conjugating hyaluronic acid (HA) with tumor-targeting ability to PPT for targeted delivery and controlled release. The HA-CO-O-PPT spontaneously assembles into nano-spherical micelles in an aqueous medium, which has outstanding serum stability and blood compatibility. The obtained prodrug micelles (HP micelles) exhibit a pH-responsive drug release mode with cumulative release reaching 81.2% due to dissociation in response to acid stimulus and have a high cellular uptake efficiency exceeding 97% as a result of HA receptor-mediated targeting.
To further optimize these HP micelles, a pH/reduction dual-responsive prodrug (HA-S-S-PPT [diameter = 98.30 nm]) was constructed via a three-step reaction protocol, wherein HA was conjugated to PPT through ester and disulfide linkages [85]. The micelles self-assembled from HA-S-S-PPT prodrug efficiently accumulate at the tumor site due to HA receptor-mediated endocytosis and exhibit a 33.1% higher cumulative release than pH-responsive micelles HA-NH-CO-PPT (diameter = 124.57 nm), reflecting the dual responsiveness to pH and reduction. HA-S-S-PPT micelles with negligible systemic toxicity achieve excellent in vivo antitumor activity with a tumor inhibition rate reaching 92%, which was significantly higher than that of pH-responsive HP micelles.
The adoption of polymeric micelles as a nanodevice for PPT administration illustrates enormous promise in terms of diminishing cytotoxicity to normal cells, while increasing toxicity to cancer cells. In addition, the polymeric micelle formulation sustained release of PPT adds to improved and prolonged therapeutic efficacy.
2.2.2 Polymer-drug conjugates (polymeric prodrugs)
Polymer-drug conjugates, also known as polymeric prodrugs, self-assemble into nanoparticles in aqueous solution, which allows for passive targeting via the EPR effect [86, 87]. The chemical bonds connecting the drug and the carrier are usually tumor-environment responsive. Disulfide and trisulfide bonds, for example, are responsive to high levels of GSH in tumor cells, while carbonate and hydrazone bonds are responsive to low pH in the tumor microenvironment [88–91]. In addition, polymeric prodrugs significantly enhance the in vivo drug stability and controllability without changing the original medicinal use of the drug [92]. In light of these findings, polymer prodrugs are also utilized to overcome PPT defects and its derivatives with strong toxic side effects and poor water solubility.
Through one-pot esterification, PPT was coupled with PEG onto an acetylated carboxymethyl cellulose (CMC) backbone to generate a PPT-PEG conjugate, which was then self-assembled into nanoparticles [93]. The conjugate with a low PPT/PEG molar ratio of 2 yielded NPs with a mean diameter of 20 nm and released PPT at approximately 5% per day in serum, while conjugates with increased PPT/PEG ratios (5 and 20) produced larger particles (30 and 120 nm, respectively) and displayed slower drug release. The 20-nm particles exhibited 2-5-fold enhanced cell killing potency and 5-20-fold increased tumor delivery compared to the larger NPs. The PPT-PEG conjugate also exhibited much higher tumor-targeted properties over healthy tissues. The 20-nm PPT-NPs had minimal toxicity compared to the larger PPT-NPs, free PPT, and standard taxane chemotherapies, and had noticeably improved efficacy against MDR-tumors in mice.
After optimizing the molar ratio of PPT-to-PEG, the PPT-PEG conjugate polymer, Celludo®, containing 23 mol% PPT (19 wt%) and 14 mol% PEG (39 wt%) was developed, then self-assembled into NPs in normal saline with a median diameter of 20.3 ± 1.8 nm and a polydispersity index of 0.1 ± 0.04 [94]. Celludo® has extended blood circulation with an 18-fold prolonged half-life (t1/2), a 9000-fold higher area under the curve (AUC) and a 1000-fold reduced clearance compared with free PPT. Furthermore, Celludo® was shown to have selective localization in lung metastatic nodules and increased the median survival to 20 d compared to 6-8 d with docetaxel and plain PPT treatment. Celludo® prolonged the median survival from 50 to 70 d in an intraperitoneal metastatic model of a human ovarian NCI-ADR/RES tumor, whereas the standard therapy (PEGylated liposomal doxorubicin) had no effect. No major toxicity was detected with Celludo® treatment.
The sensitive linkers in almost all PPT-prodrugs are ester bonds, which result in slow and incomplete drug release. A near-infrared (NIR) theranostic prodrug (DCM-S-PPT) and its amphiphilic copolymer (methoxy-PEG [mPEG]-DSPE)-encapsulated NPs was designed and synthesized, in which PPT was conjugated with dicyanomethylene-4H-pyran (DCM) via a thiol-specific cleavable disulfide bond [95]. DCM-S-PPT has much lower in vitro cytotoxicity against healthy human cells (293T) and excellent antitumor activity to Hela cells because GSH can activate DCM-S-PPT, which results in PPT release. By encapsulating DCM-S-PPT in the hydrophobic part of mPEG-DSPE, the assembled mPEG-DSPE/DCM-S-PPT micelles acquired aqueous solubility and biocompatibility, exhibiting better tumor targeting than DCM-S-PPT alone, which was due to the EPR effect.
Similarly, an innovative GSH-responsive PPT conjugate was produced by coupling PPT to an anti-mitotic cell-penetrating peptide (PRA) via a disulfide linkage [96]. The prepared PPT-PRA conjugate was self-assembled into a vesicle in water, then loaded with another anti-cancer drug for synergistic drug delivery. The vesicle was later modified with folic acid (FA) for better cancer cell targeting. The FA-modified drug-loaded vesicle not only overcame the poor water solubility and side effects of PPT, but also exhibited targeted toxicity and synergistic therapeutic effects.
An amphiphilic conjugate prodrug (MTX-SS-PPT) was constructed in which hydrophilic methotrexate (MTX) and PPT were linked via a disulfide bond to generate a novel reduction-responsive PPT delivery system [97]. The conjugate self-assembled into spherical nanoaggregates in an aqueous solution and self-delivered to tumor tissues, where over-expressed GSH broke the disulfide bonds and released free PPT and MTX. Subsequently, the released MTX in turn enhanced the targeted capability to FA receptor-positive cells ( Figure 6 ). In vitro and in vivo experiments indicated that the nanodrug effectively improved biocompatibility and mitigated the toxicity of PPT.
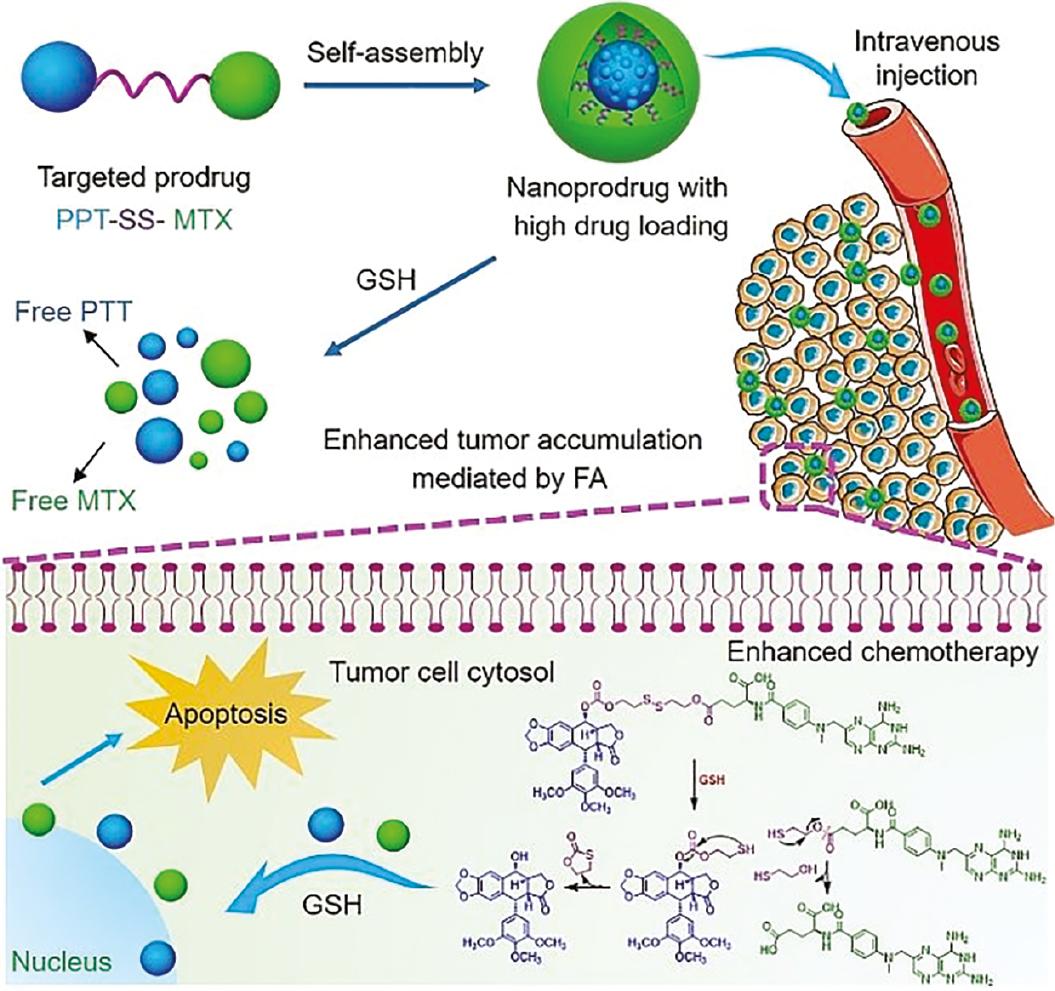
PPT prodrug nanoaggregates with high drug loading and tumor-specific drug release for improving the efficacy of chemotherapy (adapted from reference [97]).
PPT was linked to PEG (n) monomethacrylate (PEGMA) via an H2O2-responsive oxalate ester bond to produce an inspirational new prodrug (PPT-PEG), which self-assembled into stable NPs (PPT-PEG NPs) [98]. PPT-PEG NPs can be activated by hydrogen peroxide, resulting in PPT release and are extremely toxic against colon carcinoma CT26 cells ( Figure 7 ). In vivo biodistribution studies demonstrated that PEGylated PPT-PEG NPs are capable of prolonging blood circulation. Intravenous injection of PPT-PEG NPs into CT26 tumor-bearing Balb/c mice resulted in enhanced therapeutic efficacy against tumors, without increased systemic toxicity.
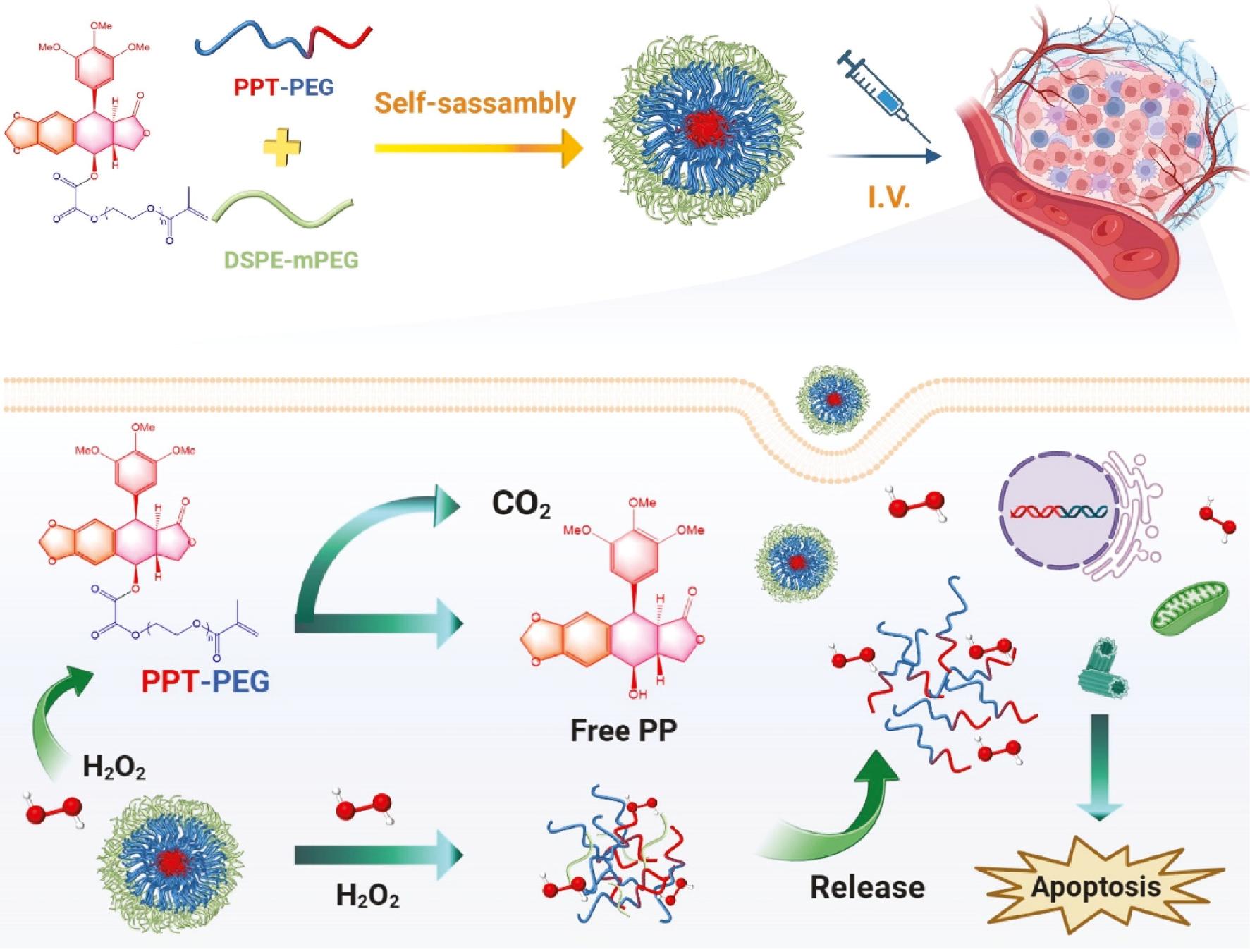
Schematic illustration of the H2O2-responsive linkage-bridged prodrug PPT-PEG NPs for anticancer drug delivery.
In the tumor cells, PPT-PEG respond to H2O2 and release the parent PPT drug through a self-sacrificing pathway, and thus enhance cancer therapy by improving drug delivery (adapted from ref. [98]).
Polyacrylic acid (PAA), one of the extremely hydrophilic, absorbent, and biodegradable polymers, was esterified with PPT to establish a water-soluble ester prodrug [99]. The podophyllotoxin-polyacrylic acid conjugate (PPC) was self-assembled into micelles at room temperature with a particle size of 215 ± 11 nm. An in vitro study demonstrated that the PPC micelles caused significantly less hemolysis (18.6 ± 2.9%) than plain PPT solution, and revealed strikingly (p <0.01) higher in vitro cytotoxicity against both sensitive and resistant human breast cancer cells (MCF-7 and MDA MB-231) after 48 h of treatment.
Hinokitiol is known to have antibacterial and anticancer activities [100]. A heterodimeric prodrug of PPT-hinokitiol linked with a dicarboxylic acid (PPT-Cn-Hinoki) was successfully produced using the reprecipitation method without using any nanocarriers. By changing the length of the carbon chain linker (n = 4, 10, and 18), the longer the length of the carbon chain linker, the smaller the size of NPs and the lower the degree of cytotoxicity [101]. Additionally, hinokitiol-modified PPT improves the stability of nano-prodrugs compared to PPT dimer.
NPs produced from homodimeric prodrugs with high drug loading potential are emerging as prospective nanomedicines [102]. Liang et al. [103] created novel ROS-sensitive PTV-NPs with high drug loading, as well as self-amplifying drug release potential by incorporating PPT dimeric prodrug and VK3 into a biocompatible polymer Pluronic F127. The resulting PTV-NPs selectively increase ROS activity in cancer cells in vitro and in vivo, which accelerates the PPT dimeric prodrug conversion to active PPT, leading to a remarkable decrease in side effects and an increase in antitumor efficacy.
Three novel PPT-fluorene methanol prodrugs (α-Fmoc-SS-PPT, β-Fmoc-SS-PPT, and γ-Fmoc-SS-PPT [FAP, FBP, and FGP, respectively]) were designed and synthesized by linking PPT to different lengths of disulfide bonds [104]. All three PPT prodrugs self-assembled into uniform NPs with high drug loading (>40%) via the one-step nano precipitation method, which not only avoids the use of surfactants and co-surfactants, but also reduces the systemic toxicity of PPT and increases the tolerated dose. Among the three prodrug (NPs, FAP, NPs containing an α-disulfide bond) had the most sensitive tumor-specific response and most rapid drug release rate, thus demonstrating the greatest in vitro cytotoxicity. In addition, three prodrug NPs showed prolonged blood circulation and higher tumor accumulation, but FAP NPs demonstrated the most potent in vivo antitumor activity.
E-selectin is considered a promising antitumor-targeted protein in tumor therapy because of differential expression in pathologic states and a strong affinity to vascular endothelial cells near the tumor when endothelial cells are stimulated by inflammatory factors [105, 106]. A PEGylated PPT-loaded E-selectin peptide conjugate was designed and prepared that further self-assembled into water soluble NPs. The released PPT may specialize in tumors via targeting endothelial cells in the tumor microenvironment, resulting in superior in vivo tumor growth inhibition, intratumorally enhanced microvessel density, and lower systemic toxicity of this NP over free PPT [107].
Taken together, these findings suggest that polymeric prodrug nanosystems not only transport PPT to the targeted site for controlled release, but also achieve synergistic effects through drug combination.
2.2.3 Dendrimers
Dendrimers are geometrically and physically comparable to colloidal particles, which are particularly noticeable in higher generations of colloidal dendrimers. The distinguishing characteristics make dendrimers suitable for a wide range of sectors, including medicine and biomedicine [108]. To minimize toxicity and enhance solubility under physiologic conditions, PPT was conjugated to a poly(amidoamine) [PAMAM] dendrimer to form a sustained-release PPT dendrimer conjugate (D-PPT) [109]. According to histologic, biochemical, and mortality investigations, 8 mg/kg of D-PPT was benign, but the same amount of free PPT was highly toxic. D-PPT therapy decreased epithelial hyperplasia and induced apoptosis by raising the Bax-to-Bcl-2 ratio and decreasing NF-kB expression according to histopathologic and immunohistochemical investigations. More notably, higher doses of the D-PPT conjugate exhibited fewer adverse effects and higher effectiveness than free PPT. Further research revealed that when mice with chemically-induced hepatocellular carcinoma (HCC) were treated with D-PPT, the histologic alterations in liver tissues were dramatically reduced. D-PPT significantly lowered the levels of inflammatory markers (IL-6 and NF-B) in serum and tissues, respectively, as well as fibrous tissue deposition in the liver [110].
Polymer-based nanocarriers are widely used in PPT delivery. Most polymeric carriers loaded with drugs self-assemble into NPs to achieve passive targeting via the EPR effect, and realize active targeting and sustained release of the drug by incorporating stimuli-responsive properties into the NPs. In fact, different polymeric NPs have strengths and weaknesses. The spherical core-shell structure in polymeric micelles facilitates entrapment of hydrophobic pharmaceuticals in the core and protects the formed micelle against aggregation. In addition, disadvantages, such as off-targeting and burst drug release, can be addressed by incorporating stimuli-responsive properties in the micelles. Polymer-drug conjugates generally exhibit better stability, lower toxicity, higher drug-loading capacity, and prolonged drug release behaviors compared to other conventional polymer-based platforms. Nevertheless, major challenges for polymeric prodrugs, such as rapid cellular internalization and ineffective delivery to cancer cells, can be improved through structure modification. The presence of an enormous amount of terminal end units in dendrimer networks allows polymeric prodrugs to be multifunctional. Development and application have been severely constrained due to the intricacy of the synthesis process and the lack of mass production methods.
2.3 Others
In addition to lipid- and polymer-based nanocarriers, several other types of nanocarriers are utilized for PPT delivery. For example, layered double hydroxides (LDHs), inorganic NPs with a unique anion exchange capability, controlled release, and payload protection, have recently gained a lot of attention for promising uses in biomedicine [111–113]. Unprecedented inorganic nanohybrids of PPT have been designed and manufactured by intercalating PPT into LDHs to form a PPT-LDH [114]. The particle size of PPT-LDH is merely 80–90 nm and the zeta potential is 20.3 mV. PPT-LDH NPs outperformed conventional PPT in in vitro and in vivo anti-tumor efficacy and more readily uptake by Hela cells. PPT delivery via LDH NPs is more efficient and sustained, and the pharmacokinetics study displays a prolonged circulation time and an increased bioavailability of PPT-LDH than free PPT, suggesting greater in vivo stability.
Cyclodextrins (CDs) are prominently suitable for functional delivery systems in the pharmaceutical industry [115, 116]. Gamma-cyclodextrin (γ-CD), because of its appropriate geometric size, was selected to prepare a PPT inclusion complex [117]. The inclusion complex formed in a 1:1 ratio with a considerable apparent stability constant Ks (4245.5 L·mol−1), which greatly improved PPT water solubility. In addition, the anticancer activity of the inclusion complex was better than the positive cis-platinum (DDP) control, which probably provided a strong probability of expanding its clinical applications.
Hydroxypropyl-β-cyclodextrin (HPβCD) features relatively high water-solubility with low toxicity and adequate inclusion ability, which was utilized to form a PPT-HPβCD inclusion complex that was shown to be cytotoxic to all investigated human tumor cell lines with an easy and environmental-friendly preparation method ( Figure 8 ) [118].
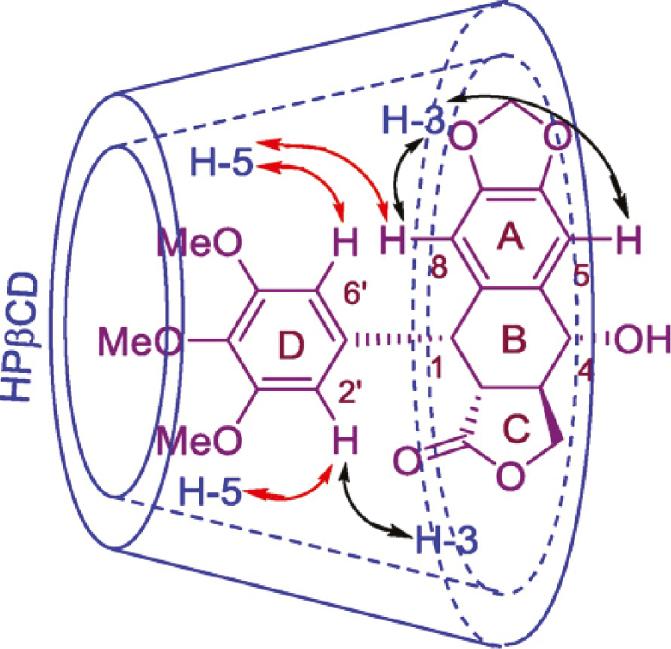
Possible inclusion mode and significant NOESY (↔) correlations of the PPT/HPβCD inclusion complex (adapted from ref. [118]).
Then, two pH-responsive inclusion hosts [mono-(6-deoxy-6-amino)-β-CDs] modified with maleamide derivatives were developed and prepared for PPT delivery, which significantly increased the water solubility of PPT by > 190- and 500-fold, respectively [119]. Additionally, the PPT/host inclusion complexes retain anticancer efficacy while significantly reducing cytotoxicity in healthy cells.
Based on previous work, a graphene oxide (GO)-based novel drug delivery system was developed to load a PPT disulfide prodrug (DCM-S-PPT) [120]. To improve biocompatibility in the physiologic solution, GO was functionalized with a 6-armed PEG. DCM-S-PPT was loaded on GO-PEG via p–p stacking and hydrophobic interactions. The loading rate reached 138%. An in vitro cytotoxicity assay showed that GO-PEG/DCM-S-PPT inhibited the proliferation of human cervical adenocarcinoma HeLa cells more than healthy human kidney 293T cells, which was attributed to the different intracellular concentrations of GSH. After intravenous injection in tumor-bearing mice, GO-PEG/DCM-S-PPT showed better antitumor activity and fewer side effects than DCM-S-PPT and free PPT.
Taken together, different nano delivery systems for PPT loading can enhance PPT water solubility, alleviate unwanted toxicities, and improve antitumor potency and targeted delivery, and some may even prolong median survival in mice. Consequently, nano delivery carriers possess great opportunities for PPT formulation preparation in comparison to conventional drug administration ( Table 1 ).
Summary of PPT-loaded nanoparticles.
| Types | Carrier material | Method | Particle size (nm) | EE% | Cell line/animal model | Characteristics |
|---|---|---|---|---|---|---|
| Liposomes | DPPC, cholesterol | Reverse phase Evaporation | 1450 | 73.8 | - | [42] |
| Soya lecithin, cholesterol | 3780 | 79.1 | - | |||
| Cholesterol to lecithin | Thin-film dispersion | 106 | 90.425 | PC3 cells | Enhanced anti-cancer and cell uptake activity [43] | |
| Succinate, GPC | Thin film | 160 | - | MCF-7, HeLa, and U87 cells; BALB/c nude mice | Improved stability and reduced side effects [44] | |
| Phosphorylcholine | Self-assemble | 182 | - | MCF-7, HeLa, and HepG-2 cells | Enhanced intracellular internalization and efficient cytotoxicity [45] | |
| PBA, tri-peptide cationic lipid | Self-assemble | 100-130 | - | H460, HL7702, and H460 tumor-bearing mice | Better tumor-targeting activity and fewer side effects [49] | |
| Deoxycholic acid, soybean phospholipid, L-cysteine | Thin membrane dispersion | 172.5 ± 67.2 | 84.47 ± 3.84 | Porcine ear skin; female ICR mice | Accumulation in the epidermis without skin irritation [51] | |
| SLNs | Tripalmitin, soybean lecithin, poloxamer | High-pressure homogenization | 73.4 | - | Full-thickness abdomen skin from a single porcine | Targeted epidermic effect and controlled release [57] |
| Tripalmitin, soybean lecithin, polysorbate 80 | 123.1 | |||||
| Stearic acid, lecithin, Myrj59 | Solvent emulsification-evaporation | 50 | 71.6 | 293T and HeLa cells | Slower release and prolonged anti-tumor activity as well as reduced side effects [58] | |
| NLCs | Lecithin, monostearin, caprylic/capric Triglycerides, poloxamer 188 | Emulsion Evaporation | 85 6 ± 10 25 | 88.56 ± 3.1 | H8 cells | Enhanced tumor-growth Inhibition and induced apoptosis activity [66] |
| Compritol® 888 ATO, Labrasol®, Cremophor® RH40, soybean phosphatidylcholine | Hot high-pressure homogenization | 106 | 88.25 | Abdominal skin from male Sprague-Dawley rats | Enhanced skin-targeting efficiency and reduced systemic toxicity [68] | |
| 219 | 93.50 | |||||
| Glyceryl monostearate, octyl/decyl acid triglyceride, lecithin, poloxamer | emulsion Evaporation and low-temperature solidification | 178.5 ± 20 | 82.9 ± 2 | VK3/E6E7 cells, female Tibetan mini-pigs | Prolonged HPV inhibition and reduced inflammatory cytokine production [69] | |
| Polymeric micelles | Stearic acid, chitosan oligosaccharide | Dialysis | 346.4 ± 6.8 | 70.2 | MCF-7, A549 and Bel-7402 | Enhanced targeted delivery and anti-tumor activity [78] |
| PLA, PVA | Solvent evaporation | 100.04 ± 17 | 17 | A549 and CHO-K1 cell lines | Increased anti-tumor activity [79] | |
| T7-peptide, PEG/mPEG, dithiodipropionicanhydride | Solvent-evaporation | 125.7 ± 2.3 118.8 ± 3.4 | 100 | MCF-7, MCF-7/ADR, A549, A549/PTX cell lines, mice bearing MCF-7/ADR xenografts | Reduced side effects and enhanced anti-tumor activity as well as increased MTD dose [80] | |
| PEG, PAH, DMA, PAMAM | Synthesis | 13.3 | - | A549/PTX, MCF-7/ADR; Kuming mice | Size-shrink property and enhanced tumor accumulation [81] | |
| MPEG-b-PLL, DMA, Cu B | Self-assemble | 70.1 ± 2.9 | - | A549, A549/PTX cells, A549/PTX tumor-bearing nude mice | Enhanced targeted delivery to MDR cells and increased MTD dose [82] | |
| HA, DCC | Self-assemble | 106 | - | MCF-7, A549, HL7702 cells, MCF-7 tumor-bearing mice | Enhanced targeting delivery and anti-tumor activity along with reduced side effects [84] | |
| HA, EDC, NHS, cystamine dihydrochloride, succinic anhydride | Self-assemble | 98.30 124.57 | - | MCF-7, A549, HL7702 cells; MCF-7 tumor-bearing mice | Enhanced targeted delivery and anti-tumor activity and reduced systemic toxicity [85] | |
| Polymeric prodrugs | PEG, CMC-Ac | Self-assemble | 20-120 | - | MDA-MB-231, PC3, EMT6 and EMT6/AR1 cells, EMT-6/AR1 tumor-bearing mice | Increased tumor-targeting and anti-tumor activity [93] |
| DCM, DSPE-mPEG | self-assemble | 90 | - | HeLa and 293T cells, Hela cell tumor-bearing nude mice | Improved solubility and tumor-targeting along with reduced toxicity [95] | |
| Anti-mitotic cell-penetrating peptide | Self-assemble | 180 | - | HepG2, HL7702, HeLa, 293T cells | Improved water solubility and enhanced targeted anti-tumor activity as well as a synergistic therapeutic effect [96] | |
| MTX, 2-hydroxyethyl disulfide | Self-assemble | 101.3 | - | 4T1, A549, and KB cells, 4T1 xenograft tumor-bearing mice | Improved tumor-targeting and reduced toxicity [97] | |
| PEGMA, DSPE-mPEG 2000 | Self-assemble | 68.0 | 83.7 | CT26 cells, NIH 3T3, CT26 tumor-bearing BALB/c mice | Enhanced targeting delivery and anti-tumor activity along with reduced systemic toxicity [98] | |
| Polyacrylic acid | Thin-film hydration, self-assemble | 215 ± 11 | - | MCF-7 and MDAMB-231, HEK-293 | Enhanced anti-tumor activity and water solubility along with reduced systemic toxicity [99] | |
| Hinokitiol, succinic anhydride, monobenzyl sebacate, octadecanedioic acid | Reprecipitation | 231 214 162 | - | KPL-4 cells | A promising template for heterodimeric prodrug [101] | |
| Vitamin K3, Pluronic F127 | Self-assemble | 70-100 | 58.3 ± 2.9 | MCF-7, NIH-3T3; BALB/c nude mice | Accelerating drug release and selectively killing cancer cells [103] | |
| 9-fluorenyl methanol,3,3’-dithioglycolic acid, 4,4’-dithioglycolic acid, DSPE-PEG | One-step nano-precipitation | 100 | - | 4T1 and 3T3 cells, 4T1 tumor-bearing mice | Prolonged blood circulation and higher tumor accumulation [104] | |
| PEG, peptide CIELLQAR | Self-assemble | 108.70 ± 0.26 | - | MCF-7, MDA-MB-231, H460, 786O, HCC827, K562, H1975, HCT-116, HUVEC cells | Improved pharmacokinetic properties and safety with reduced toxicity [107] | |
| Dendrimers | Poly(amidoamine) | Self-assemble | 8-12 | - | A549 cells, DMBA-TPA induced tumor-bearing mice | Reduced side effects and enhanced anti-tumor activity [109] |
| Others | Mg(NO3)2·6H2O, Al(NO3)3·9H2O, tyrosine | Ion-exchange process | 80-90 | - | Hela cells, Hela tumor-bearing mice | Prolonged and enhanced anti-tumor efficacy [114] |
| γ-CD | Solvent-evaporation | - | - | HCT116, A549, HepG2, and K562 cell lines, male Sprague-Dawley mice | Improved water solubility and anti-tumor activity along with reduced toxicity [117] | |
| HPβCD | Solvent-evaporation | - | - | HL-60, SMMC-7721, A549, MCF-7, and SW480 cell lines | Increased water solubility and environmental-friendly preparation [118] | |
| N-Maleic anhydride-Mono-(6-deoxy-6-amino)-β-CD, N-1,2-dicarboxylic-Cyclohexene anhydride-Mono-(6-deoxy-6-amino)- β-CD | Saturated aqueous solution method and physical mixing | - | - | H1299, Hela, Huh7, and 293T | Substantial increase in water solubility and reduced toxicity to normal cells [119] | |
| GO, 6-armed PEG, DMC | Simply mixing | 220 | - | HeLa, 293T cells, and HeLa tumor-bearing mice | Loading rate reached 138%, improved tumor-targeting and reduced side effects [120] |
3. CHALLENGES AND FUTURE PERSPECTIVES
Drugs derived from natural products have been widely employed in the treatment of many major diseases, such as cardiovascular diseases, malignant tumors, and immunologic and infectious diseases [121–124]. PPT is widely recognized as a broad-spectrum pharmacologically active molecule with remarkable antitumor activity through multiple interaction mechanisms [125]. Several PPT derivatives, particularly etoposide and teniposide, have already been used in clinical cancer therapy, demonstrating their potency as chemotherapy agents [126, 127]. However, oral or injectable administration is not an option due to its severe systemic toxicity, poor solubility, and low tolerable dose.
Nano-delivery and structural modification are currently the primary approaches and strategies to break through the bottleneck of active monomer components in the clinic [128–130]. Various nanotechnology platforms in the field of biomedicine, spanning both diagnostics and therapy, have received significant attention in recent years, presenting greater prospects for the therapeutic application of PPT. For one thing, PPT could be encapsulated into nano-carriers to get increased aqueous solubility, better stability, enhanced cellular uptake, environment-responsive release, and combination treatment. For another, PPT can be conjugated onto the nanocarriers to exert compelling inhibitory effects against tumor cells, or even act as a ligand to mediate the tumor-targeting delivery for cancer therapy, showing enhanced toxic effects to cancer cells.
To date, nanocarriers employed for PPT delivery have predominantly been lipid- and polymer-based origin, coupled with some inorganic carriers and cyclodextrin inclusions. Despite superior biocompatibility and degradability, among the primary obstacles for lipid-based carriers are particle size control and reproductivity in mass manufacturing. Furthermore, due to the susceptibility of drug efflux and “burst release” with lipid-based NPs, the key to success is adopting innovative strategies to develop stimulus-responsive functional carriers for targeted delivery and sustained release. Polymeric nanocarriers are characterized by self-assembly and excellent stability, as well as comparatively high drug loading, enhanced water solubility, and prolonged blood circulation duration. Therefore, the design and development of multifunctional polymeric NPs for improved targeting ability and controlled release of PPT via multiple stimuli-response behaviors is increasingly hailed by pharmaceutical researchers.
Despite heroic attempts to develop PPT-loaded nano-formulations for improved solubility and targeted distribution, few formulations have entered clinical trials, let alone reached the market. PPT delivery continues to confront multiple challenges [131, 132]. First, to effectively deliver PPT to targeted tissues and achieve sustained controlled release with negligible systemic toxicity, it is critical to design active targeting, tumor microenvironment-responsive, and biomimetic targeted nano-drug delivery devices for PPT loading. Second, the clinical effectiveness of these nanocarriers is dependent on crucial criteria, such as NP size, physical features, drug loading efficiency, drug release potential, and most importantly, carrier toxicity. Third, it is crucial to develop a feasible means for the mass production of nano-formulations with high batch-to-batch reproductivity. Furthermore, preclinical animal models fail to anticipate clinical outcomes with sufficient accuracy. Mouse models fall short of adequately replicating human tumors, thus widening the gap between preclinical and patient data. As a result, verifiable changes in pharmacokinetics and biodistribution are required to improve patient outcomes. Given all of these findings, the question of how to choose acceptable chemical agents to conjugate with PPT physically or chemically for the translation of PPT nano-preparations from bench-to-bedside remains a critical concern.
4. CONCLUSION
Natural products, such as PPT, have become extremely important in the prevention and treatment of various diseases; however, the poor solubility and bioavailability of natural compounds hinder therapeutic practicality. As one of the most promising chemotherapeutic drugs, PPT has been designed for nano-preparations in a more rational, efficient, and functional manner. Enhanced safety, target tissue accumulation, efficacy, and reproducibility largely determine the clinical success of PPT. Given the benefits of nanotechnology and the bioactivities of PPT, we believe it will become an ongoing research priority in the near future. PPT-involved nanosystems deserve in-depth research. The benefits of more multifunctional techniques, including combination therapy, targeted therapy, and the integration of diagnostics and medicines in one nanosystem, will assist PPT in achieving potential clinical outcomes. Additionally, continued multidisciplinary collaboration and rational design will also accelerate the clinical translation of PPT in the forthcoming year.