
Natural products are an important source for drug discovery [1, 2]; however, many natural products are not suitable for use as drugs due to issues, such as poor solubility, stability, or toxicity. In addition, the complex molecular structures of these natural products make it difficult to modify them in a straightforward manner. As a result, the utilization and advancement of drugs originating from natural products frequently encounter serious limitations [3–5]. Among the limitations, indolizidine scaffolds represent a group of pentacyclic natural products [6–9]. These characteristic skeletons have been reported to exhibit a wide range of biological activities, including anti-inflammatory, anti-cancer, anti-asthmatic, and immunosuppressive effects, as well as antiviral activity against SARS-CoV-2. Tylophorine serves as a representative example of these diverse scaffolds [10–12].
This class of alkaloids has long-standing drawbacks, including severe central nervous system (CNS) side effects, low water solubility, and low metabolic stability, which severely limit its applications. Tylocrebrine, a similar natural product to tylophorine, was shown to be unsuitable in anti-cancer clinical trials in 1966 due to CNS toxicity [13]. To address these challenges, various strategies have been used to modify tylophorine. Specifically, the introduction of hydrophilic substituents [14], incorporation of quaternary ammonium salts [15], or preparation of uncyclized E-ring tylophorine [16] have been explored to enhance stability or aqueous solubility. Although these methods have made significant progress, reduced anti-proliferation activity has often occurred.
The gem-dimethyl group is an important and unique medicinal chemistry structural motif. Incorporation of the gem-dimethyl group into drug molecules provides benefits, such as stability, solubility, pharmacokinetics, and biological activity. This versatile functional group can be strategically introduced into a variety of compounds to optimize drug-like properties, which improves the likelihood of success in the drug discovery process [17–20]. One major advantage of incorporating gem-dimethyl groups into drug candidates is the ability to improve pharmacokinetic properties. Additionally, gem-dimethyl groups protect vulnerable sites from metabolic degradation, which prolongs compound half-life and increases overall stability [21]. Gem-dimethyl groups also have a crucial role in modulating the potency of a drug candidate [22]. Moreover, gem-dimethyl groups help optimize the conformation of a molecule, which can positively impact binding affinity and overall potency [23]. For the above reasons, the gem-dimethyl group has been utilized in the development of different therapeutic agents, including kinase inhibitors [24], transcription factor inhibitors [22], and β-lactamase inhibitors [25], to improve the drug-like properties and increase the likelihood of successful clinical development ( Scheme 1 ).

A structure-activity relationship study of tylophorine demonstrated that the rigid phenanthrene structure is a prerequisite for high anti-cancer activity [13, 26, 27]. We reasoned, however, that this moiety can lead to strong intermolecular π-π stacking interactions, which results in poor aqueous solubility [28]. To address this issue, we strategically designed gem-dimethyl-tylophorine analogues, which are molecules with two methyl groups at the benzylic position of tylophorine. The steric hindrance effect of the methyl groups alters the molecular planarity, thereby potentially disrupting the intermolecular π-π interactions and improving aqueous solubility. Additionally, the increased hydrophilicity of the analogues may hinder the ability to cross the blood-brain barrier (BBB), potentially mitigating CNS side effects. Furthermore, the occupied benzylic position also enhances metabolic stability, thereby improving bioavailability ( Scheme 2a ). To test this hypothesis, we performed molecular simulations. As illustrated in Scheme 2b , the spatial conformation of the tylophorine backbone has a planar structure. The presence of the gem-dimethyl group introduces structural deviations from planarity. We hypothesized that this observation provide insight into the disruptive effects of the gem-dimethyl group, ultimately enhancing the drug-binding properties of tylophorine.

In the current study we present the design, synthesis, and evaluation of tylophorine gem-dimethyl analogues. We identified several lead compounds derived from tylophorine that exhibited excellent anti-cancer activities and significantly improved drug-like properties compared to tylophorine.
In our previous work we developed a novel photoredox-catalyzed cascade carboamination reaction that was successfully used in the synthesis of tylophorine and its gem-dimethyl analogues [29]. The diverse biological activities exhibited by tylophorine served as a source of inspiration for the discovery of rapid synthetic routes and novel bioactive molecules. Thus, we prepared compounds 1a-3a and 1b-3b ( Figure 1 ) using our new synthetic method and performed preliminary biological activity tests to validate the accuracy of our approach. The MTS or CCK-8 assay was used to assess growth-inhibitory activity of these analogues against various cancer cell lines using doxorubicin and tylophorine as reference compounds ( Table 1 ). Several gem-dimethyl-tylophorine analogues demonstrated potent anti-proliferative activity against a wide range of human refractory cancer cells. Notably, compound 1b exhibited higher potency than tylophorine against the tested cancer cell lines, which was also observed when compared to doxorubicin in most cases. Compound 2b displayed outstanding anti-proliferative activity, with IC50 values in the low nanomolar range and potency 2-10 times greater than tylophorine or doxorubicin in some cell lines (e.g., A549, H460, and Ramos). Moreover, compound 2b exhibited significant anti-proliferation effects on A549 (a human lung adenocarcinoma cell line) and MDA-MB-231 cells (a human breast cancer cell line) with IC50 values < 20 nM. While compounds 1a, 3a, and 3b did not exhibit anti-proliferative activity against the tested cancer cell lines, compounds 1a, 3a, and 3b provided valuable insight into the structure-activity relationship of tylophorine, which contributed to our understanding of target protein recognition. The promising anti-cancer activity of compounds 1b and 2b showed that gem-dimethyl-tylophorine analogues, enabled by this novel approach, could serve as potential hit compounds for further lead optimization.

Anti-proliferative activities of gem-dimethyl-tylophorine analogues.
| Cell line | IC50 (nM)
a
| ||||
|---|---|---|---|---|---|
| Doxorubicin | Tylophorine | 1b | 2b | 1a-3a, 3b | |
| A549 b | 51.3 | 116.2 | 27.7 | 10.4 | >500 |
| K562 c | 71.3 | 95.0 | 57.7 | 30.2 | >500 |
| Ramos d | 111.0 | 86.6 | 75.8 | 30.6 | >500 |
| HBL-1 e | 49.2 | 81.9 | 50.9 | 29.0 | >500 |
| RKO f | 91.4 | 42.8 | 56.1 | 33.8 | >500 |
| MDA-MB-231 g | 85.0 | 41.5 | 35.8 | 19.7 | >500 |
| H460 h | 145.5 | 160.4 | 51.1 | 34.7 | >500 |
| HeLa i | 28.7 | 45.6 | 25.7 | 29.5 | >500 |
aAll values are the mean of three experiments; bHuman lung adenocarcinoma cell line; cHuman chronic myeloid leukemia cell line; dHuman Burkitt’s lymphoma cell line; eHuman diffuse large B-cell lymphoma cell line (BTK C481S mutant); fHuman colon cancer cell line; gHuman breast cancer cell line; hHuman lung cancer cell line; iHuman cervical cancer cell line.
Based on these findings, we initiated the design and synthesis of a new analogue, compound 4b, in which the C-6 methoxy group was replaced with a hydroxyl group ( Figure 2a ). Compound 4a was obtained with a total yield of 33% through a catalytic carboamination reaction, followed by deprotection of the Bn group and reduction to form compound 4b. The growth-inhibitory activities of compound 4b were subsequently assessed against K562 (a human chronic myeloid leukemia cell line) and MDA-MB-231 cells (a human breast cancer cell line), with compound 2b used as the control. Compound 4b had outstanding anti-proliferative activity with IC50 values < 10 nM. Moreover, a notable 4-6-fold increase in potency was observed compared to compound 2b against both K562 and MDA-MB-231 cells. Even in HBL-1 cells, which exhibit resistance to ibrutinib (a human diffuse large B-cell lymphoma cell line [BTK C481S mutant]; ibrutinib GI50 = 706.0 nM) [30], compound 4b demonstrated strong inhibition of cell growth, with an IC50 value of approximately 7.8 nM. Considering the lack of effective drugs for treating ibrutinib-resistant non-Hodgkin’s lymphoma, the potent activity of compound 4b suggests its potential for further development as a therapeutic option for this unmet medical need in the future ( Figure 2b–2d ).
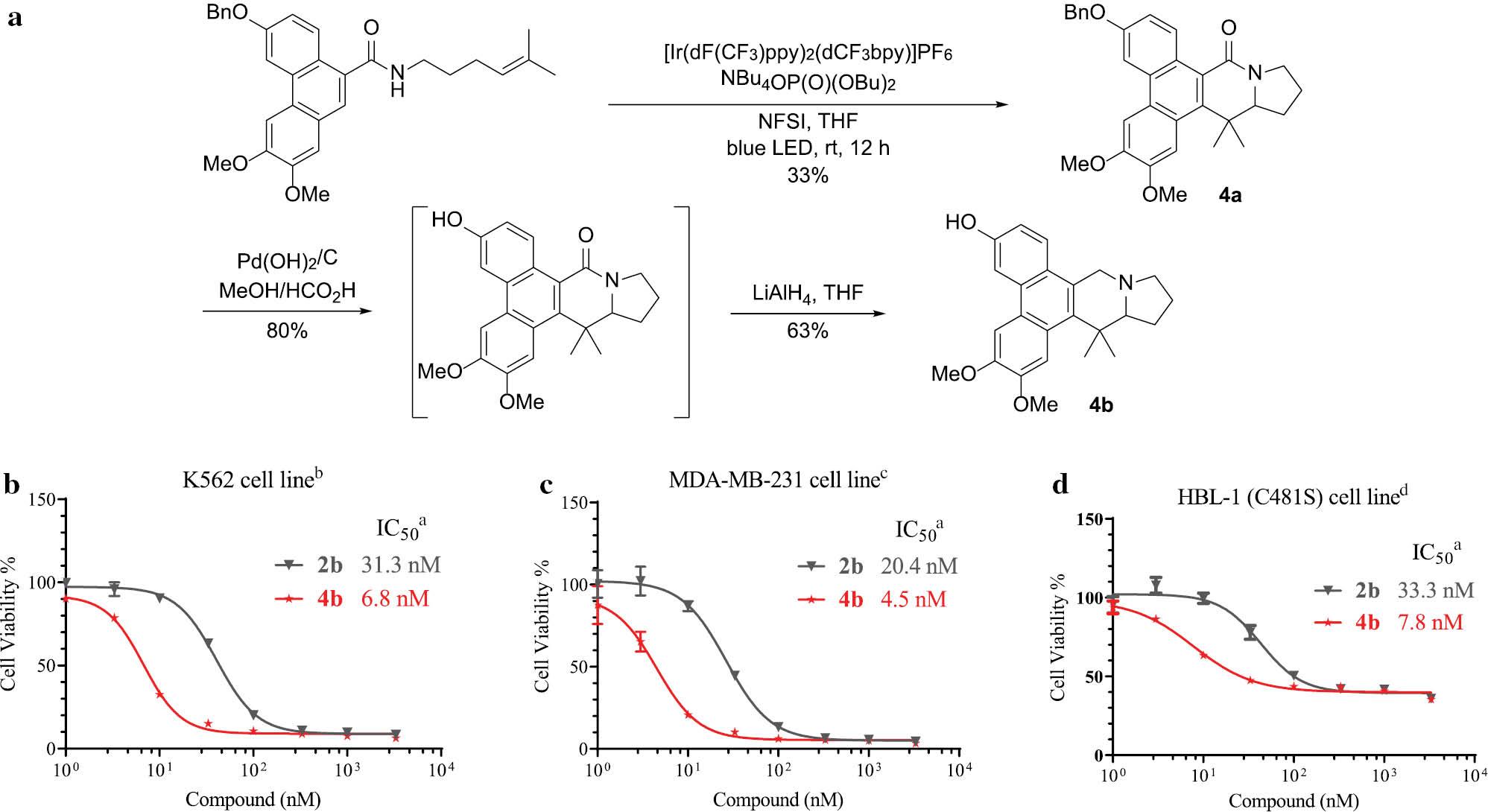
Design, synthesis, and biological tests of 4b.
aAll values are the mean of at least three experiments. bHuman chronic myeloid leukemia cell line; cHuman breast cancer cell line; dHuman diffuse large B-cell lymphoma cell line (BTK C481S mutant).
Subsequently, we performed tests on compound 4b using primary tumor cells derived from clinical patients [31] and compared the effects to paclitaxel and epirubicin. Remarkably, compound 4b exhibited excellent inhibition of tumor cell proliferation. Even in primary tumor cells resistant to paclitaxel or epirubicin, compound 4b displayed significant anti-tumor activity ( Figure 3 ).
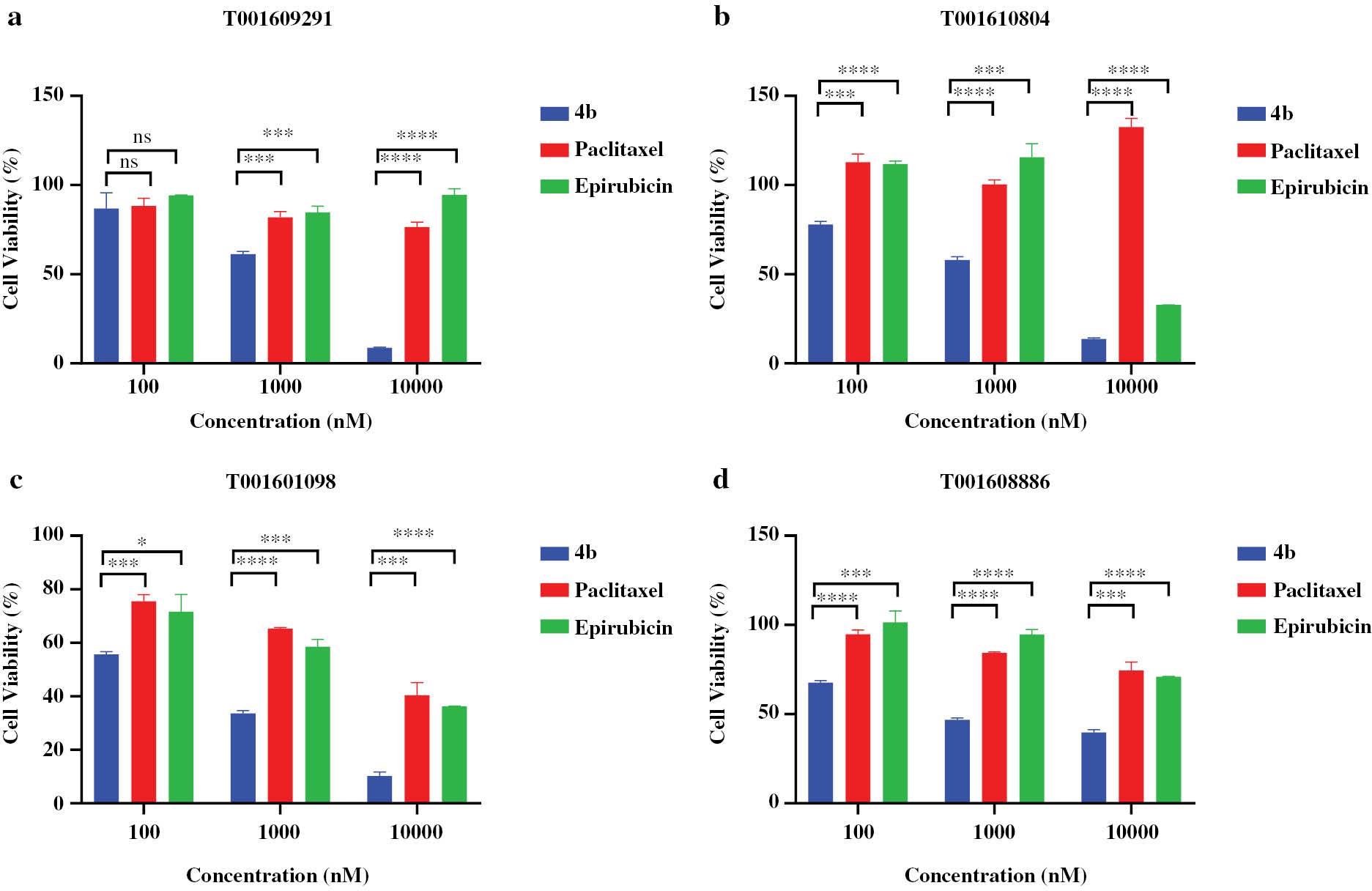
Biological tests of compound 4b in various human primary colon tumor cell lines.
All values are the mean of at least three experiments; statistical significance denoted as *P < 0.05; **P < 0.01; ***P < 0.001, ****P < 0.0001; ns, not significant.
We then successfully separated the enantiomers of compound 4b and evaluated the anti-proliferative activities. The (-)-enantiomer of compound 4b demonstrated higher potency against the tested cancer cell lines compared to compound 4b, whereas the (+)-enantiomer of compound 4b exhibited only moderate anti-proliferative activity ( Table 2 ).
Furthermore, we performed a general toxicity test on normal human bronchial epithelial cells (Beas-2b cell line). Compared to doxorubicin (IC50 = 35.3 nM), tylophorine and compounds 1b, 2b, 4b, (-)-4b, and (+)-4b exhibited significantly lower anti-proliferative activity against the tested normal cell line, as indicated by the higher IC50 values ( Table 3 ) [32]. These findings suggest a potentially improved therapeutic window for the newly derived gem-dimethyl-tylophorine analogues. Although phenanthroindolizidine alkaloids, such as tylophorine, have diverse biological activities, the poor solubility has severely limited clinical use. To address this issue, we performed solubility tests comparing tylophorine to our newly synthesized derivative, compound 4b. As depicted in Table 3 , the solubility of tylophorine was < 1 mM, whereas compound 4b exhibited a solubility value > 500 mM. Notably, the aqueous solubility of 4b in phosphate-buffered saline (PBS) was measured to be > 200 μg/mL, which was significantly higher than tylophorine (<10 μg/mL). The improved solubility of compound 4b may reduce the tendency to cross the BBB, thereby potentially minimizing CNS side effects ( Table 3 ).
Toxicity and solubility tests of the gem-dimethyl-tylophorine analogues.
| Compound | Toxicity, IC50 (nM)
a
| Solubility | |
|---|---|---|---|
| Beas-2b b | DMSO | Aqueous solution c | |
| Doxorubicin | 35.3 | ||
| Tylophorine | 627.9 | < 1 mM | < 10 ug/mL d |
| 1b | 828.8 | ||
| 2b | 614.0 | ||
| 4b | 388.7 | > 500 mM | > 200 ug/mL |
| (-)-4b | 314.5 | ||
aAll values are the mean of at least three experiments. bHuman bronchial epithelial cell line; cAqueous solubility was tested in phosphate buffered saline, (PBS [pH = 7.2−7.4]); dTested solubility data agreed with the literature reported value [15].
Furthermore, while tylophorine is easily decomposed in organic solvents [15], our gem-dimethyl-tylophorine analogue, compound 4b, demonstrated no detectable degradation as observed by 1H NMR analysis after at least 1 week, indicating increased stability ( Figure 4 ). This was an encouraging finding, suggesting that our approach offers a promising strategy for enhancing the pharmacologic and drug-like properties of phenanthroindolizidine alkaloids.

In summary, the gem-dimethyl group represents a valuable medicinal chemistry structural motif, offering potential benefits in terms of pharmacokinetic properties, potency, selectivity, and metabolic stability. We synthesized a series of tylophorine derivatives using a novel photoredox promoted reaction. Among the tylophorine derivatives, compound 4b exhibited remarkable anti-proliferative activities with IC50 values < 10 nM against 8 human cancer cell lines, including K562, MDA-MB-231, and C481S HBL-1, as well as primary tumor cell lines from patients. Compound 4b demonstrated a remarkably strong inhibitory effect (7.8 nM) against ibrutinib-resistant C481S HBL-1 tumor cells, especially for C481S ibrutinib-resistant non-Hodgkin’s lymphoma, which is due to a cysteine–to–serine mutation at position 481. Because there is currently no available therapeutic drug for ibrutinib-resistant non-Hodgkin’s lymphoma treatment, compound 4b should undergo further development for addressing this challenging medical problem in the future. Furthermore, compound 4b exhibited significant improvements in solubility and stability compared to tylophorine. Hence, there is hope that compound 4b can effectively address the drawbacks associated with tylophorine, thereby expanding its utilization and clinical application. Taken together, we believe our study may pave the way for developing new therapeutic agents for potential drug-resistant cancer treatment. With the strategic incorporation of gem-dimethyl groups, researchers can optimize drug candidates for improved efficacy and safety, ultimately contributing to the development of new and innovative therapeutics.
