
1. INTRODUCTION
During the past few years, the immune response has been found to play an essential role in combating tumor progression, metastasis, and recurrence [1]. Activation of the immune system has become central to cancer treatment. The cellular stimulator of interferon genes (STING) signaling pathway, a crucial immune-regulation switch, connects cytosolic deoxyribonucleic acid (DNA) sensing to host immune defense [2]. Notably, STING not only induces dendritic cell (DC) maturation, and thus activates CD8+ T cells via antigen cross-presentation [3], but also leads to the production of proinflammatory cytokines (e.g., tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6)), and converts “cold” tumors (displaying low response rates to immunotherapy) into “hot” tumors (with enhanced sensitivity to immunotherapy). STING is now considered a strategy to introduce innate immunity as well as adaptive immunity, thus ultimately enhancing antitumor activities [4, 5].
In recent years, STING-based therapeutics have been used in preclinical research and clinical practice [6]. However, owing to several challenges, none have yet been approved by the Food and Drug Administration. First, because of their negative charge and highly polar nature, their access to the plasma membrane is hindered [7]. Moreover, some STING agonists (e.g., exogenous DNA and cyclic dinucleotides) are easily hydrolyzed by nucleases (e.g., deoxyribonuclease I (DNase I), ecto-nucleotide pyrophosphatase 1 (ENPP 1), and deoxyribonuclease III (DNase III)), and rapidly cleared [8, 9]. Therefore, the responsivity to STING-based therapeutics is low. The duration of STING stimulation is also a critical concern. Acute and localized STING activation generally triggers an appropriate immune response to defeat diseases, but chronic STING signaling may elicit inflammation-associated diseases. The magnitude of STING signaling is another important consideration. Moderate activation of STING signaling in T cells maintains the CD8+ T cell stemness, but hyperactivation of STING can drive antiproliferative and apoptotic signaling in T cells. Therefore, precise drug delivery and accurate STING modulation remain essential.
Encouragingly, nanotechnology has emerged as an effective approach for drug delivery and immunomodulation [10]. Because of their nanoscale sizes, nano-formulations have enabled multiple advances and inspired the development of STING-based therapeutic strategies. A targeting and controlled-release platform for precise control of the magnitude and kinetics of targeted STING activation can be achieved through nanotechnology. Nanoparticles facilitate targeted accumulation of STING agonists in tumor tissues and local draining lymph nodes through drainage from the interstitial space [9, 11, 12]. In addition, polymers or liposomes not only protect cyclic dinucleotides (CDNs) from hydrolases but also help STING agonists to deliver into cytoplasm by membrane fusion and endosomal escape. Moreover, cytosolic DNA induced by classical cancer interventions leads to iatrogenic STING activation, which can be strengthened by synergistic therapies, thus eliciting an effective anti-cancer immune response. To date, STING activation strategies have promoted a revolution in immunotherapy [13, 14].
Overall, the STING pathway has become a mainstay in boosting cancer treatment. However, the safety and efficacy of delivery of STING-related therapeutic ingredients remain challenges to be addressed, for which nanotechnology may provide promising solutions. Herein, we summarize recent advances in STING modulation in cancer treatment. The enhancement of nanotechnology-based strategies in delivery, activation, and applications in STING modulation are outlined ( Figure 1 ). Moreover, we discuss future prospects and challenges. We expect that continuing research in nanotechnology-based applications of STING activation will lead to the clinical application of STING agonists. Therefore, this review article comprehensively discusses nanotechnology-based STING activation for achieving anti-tumor immune responses, and recent advances in their biomedical applications in detail ( Figure 2 ).
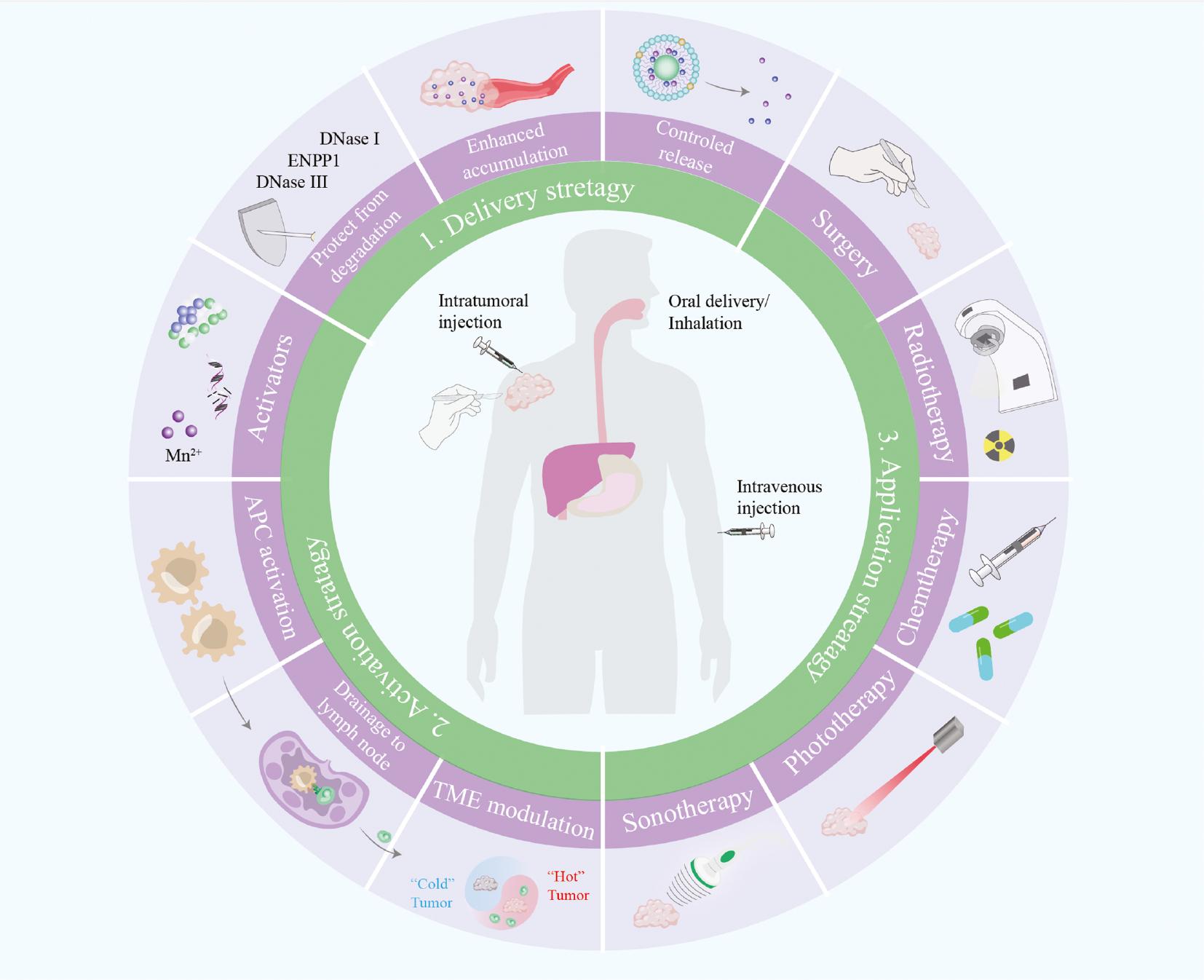
Nanotechnology-based delivery strategies, activation strategies, and application strategies in STING-mediated cancer treatment.
Nanotechnology-based strategies are used to address the issues in efficient STING activation. Delivery strategies include novel administration routes, protection from degradation, enhanced accumulation, and controlled release in tumor sites. Activation strategies include the development of novel agonists, antigen-presenting cell (APC) activation, enhanced drainage to lymph nodes, and tumor microenvironment (TME) modulation. Application strategies include surgery, radiotherapy, chemotherapy, phototherapy, and sonodynamic therapy. APC: antigen-presenting cell; TME: tumor microenvironment.
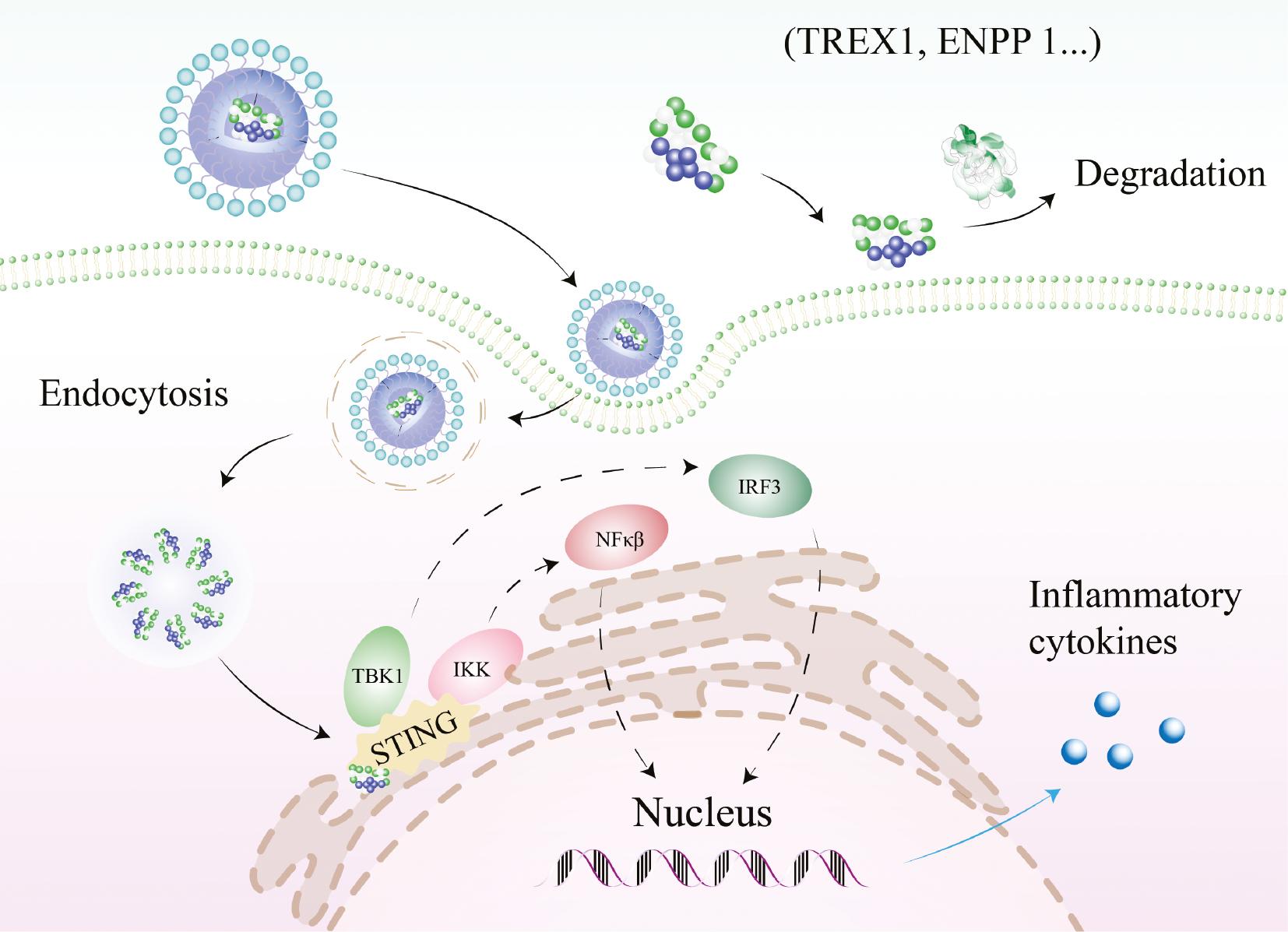
Overview of the interaction between STING signaling and agonists.
First-generation STING agonists are mainly CDNs, are easily hydrolyzed by nucleases (3′ repair exonuclease 1 (TREX1), ENPP 1, etc.), and have hindered access to the plasma membrane. Nanotechnology-based novel agonists and strategies boost intracellular STING activation. Downstream signaling pathways, including activated tank-binding kinase 1 (TBK1) and IKK (inhibitor of nuclear factor-κB kinase), lead to the recruitment and phosphorylation of IRF3 and NF-κB. Finally, STING activation enables enhanced production of inflammatory cytokines and triggers an anti-cancer immune response. CDN: cyclic dinucleotides; TBK1: Tank-binding kinase 1; IRF3: interferon regulatory factor 3; NF-κB: nuclear factor-κB.
2. STING AND CANCER TREATMENT
STING, after its discovery, was considered to be involved in host immune defense against foreign microorganisms [15, 16], whereas dysfunctional STING signaling was believed to contribute to the pathogenesis of various human diseases [17, 18]. STING provides cytosolic immune surveillance and stimulates an innate immune response to address potential threats. During the past few years, immunotherapy has reshaped antitumor strategies and achieved superior outcomes. Thus, STING has attracted researchers’ attention for its crucial role in immune regulation.
In general, the accumulation of double stranded DNA (dsDNA), including extrinsic DNA (e.g., from tumor-drive exosomes or viral infection) or intrinsic DNA (e.g., mitochondrial or nuclear), in the cytosol is considered to initiate STING activation. Traditional anticancer interventions, such as radiotherapy, chemotherapy, and phototherapy, lead to the leakage of broken DNA into the cytosol. After cytosolic dsDNA is detected by mammalian cells, the production of cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) (cGAMP) by cyclic GMP-AMP synthase (cGAS) is triggered. Subsequently, cGAMP is bound by STING and activates the transcription factor interferon regulatory factor 3 (IRF3) and induction of type I interferon (IFN), including IFN-β [2]. The activation of signals downstream of STING pathways, the stimulation of the immune response, and the release of pro-inflammatory cytokines are believed to be beneficial for cancer eradication. Therefore, by releasing dsDNA into the cytosol, conventional interventions enable STING activation and ultimately induce effective anti-cancer immunity.
Furthermore, direct STING activation by agonists also triggers an effective anti-cancer response. The compound 5,6-dimethylxanthenone-4-acetic acid (DMXAA) has shown STING activation activity in mice. Several phase I clinical trials have indicated that high doses of DMXAA alone can be used directly treat tumors [19, 20]. More importantly, DMXAA has been demonstrated to exhibit potent STING activation ability in preclinical studies [21, 22], and has been validated in immunotherapy applications [23]. Meanwhile, a phase I trial, NCT00863733, has shown good safety of DMXAA without myelosuppression, thus indicating that this treatment may be favorable in combination with chemotherapy [24, 25]. However, further clinical trials were stopped because of DMXAA’s poor efficacy and substantial adverse reactions [26], owing to the dynamic structural differences between human STING (hSTING) and murine STING (mSTING), and DMXAA only can combine to mSTING [27].
Single-nucleotide polymorphisms in the STING protein lead to distinct human STING (hSTING) isoforms. The five most prominent haplotypes of hSTING are WT (R232), HAQ (R71H, G230A, R293Q), REF (R232H), AQ (G230A, R293Q), and Q (R293Q) [28]. Different structures contribute to agonist-specific differences in the recognition and activation of the various STING isoforms. Endogenous 2′3′-cGAMP activates all five major hSTING variants, but not all bacterially derived CDNs can activate the hSTINGREF or hSTINGQ isoforms. Moreover, hSTINGHAQ exhibits lower instrinsic IFN-I (compared with other haplotypes of hSTING) and nuclear factor kappa-B (NF-κB) activity. These findings indicate a crucial design consideration for STING activation strategies for clinical translation.
3. FIRST-GENERATION STING AGONISTS
Advances in understanding have paved the way to efficient immunotherapy. Before the discovery of STING, CDNs had long been used as adjuvants to strengthen the immune response. After CDNs were reported to activate cGAS, pharmaceutical industries began clinical trials of CDNs’ application in cancer treatment ( Table 1 ). As early as 2016, phase I clinical trials of ADU-S100 were conducted [29]. However, owing to the poor phase I clinical data of ADU-S100 in combination with the Novartis PD-1 antibody spartarizumab in 2019 [30], Novartis terminated the collaborative development of ADU-S100, and Aduro subsequently stopped phase II clinical trials. Another example was MK-1454 monotherapy, for which complete or partial responses were not observed [31].
Clinical trials for STING agonists.
| ID | Drug | Agonist | Administration | Pharmaceutical industry | Stage of clinical trial | Initial time | Status |
|---|---|---|---|---|---|---|---|
| NCT03956680 | BMS-986301 | CDN | i.t. | Bristol Myers Squibb Company | Phase I | 2019.03.26 | Active, not Recruiting |
| NCT04109092 | E7766 | CDN | Intravesical administration | Eisai Inc. | Phase I | 2020.02.13 | Completed |
| NCT04144140 | E7766 | CDN | i.t. | Eisai Inc. | Phase I | 2020.02.24 | Completed |
| NCT04220866 | MK-1454 | CDN | i.t. | Merck Sharp & Dohme LLC | Phase II | 2020.03.14 | Completed |
| NCT03249792 | NCT03249792 | CDN | i.t. | Merck Sharp & Dohme LLC | Phase I | 2017.09.20 | Completed |
| NCT04592484 | ExoSTING | CDN | i.t. | Codiak BioSciences, Inc | Phase I/II | 2020.09.15 | Completed |
| NCT00863733 | DMXAA | CDN analog | i.v. | Cancer Research UK | Phase I | 1996.05 | Completed |
| NCT03589339 | NBTXR3, nivolumab, pembrolizumab | dsDNA | i.t. | Nanobiotix | Phase I | 2019.01.16 | Recruiting |
| NCT05321940 | STAVs | STING-dependent adjuvants | Undocumented | Juan C. Ramos, MD | Phase 1 | 2023.08.15 | Not yet Recruiting |
| NCT05070247 | TAK-500 | CDN | Undocumented | Takeda | Phase 1 | 2022.04.26 | Recruiting |
| NCT04420884 | TAK-676 | CDN | i.v. | Takeda | Phase 1 | 2020.07.22 | Recruiting |
| NCT04020185 | IMSA101 | CDN | i.t. | ImmuneSensor Therapeutics Inc. | Phase 1/II | 2019.09.23 | Recruiting |
| NCT05846646 | PULSAR-ICI ± IMSA101 | CDN | i.t. | ImmuneSensor Therapeutics Inc. | Phase II | 2023.06.28 | Recruiting |
| NCT02675439 | ADU-S100 ± ipilimumab | CDN | i.t. | Chinook Therapeutics, Inc. | Phase I | 2016.04.28 | Terminated |
| NCT03937141 | ADU-S100 | CDN | i.t. | Chinook Therapeutics, Inc. | Phase II | 2019.08.26 | Terminated |
| NCT04147234 | BI 1387446 | CDN | i.t. | Boehringer Ingelheim | Phase I | 2020.03.09 | Active, not Recruiting |
| NCT04096638 | SB 11285 | CDN | i.t. | nvoX Pharma Limited | Phase I | 2019.09.23 | Recruiting |
| NCT04609579 | SNX-281 | Small-molecule activator | Undocumented | Stingthera, Inc | Phase I | 2020.11.18 | Recruiting |
| NCT05424380 | GSK-3745417 | Small-molecule activator | Undocumented | GlaxoSmithKline | Phase I | 2022.09.20 | Recruiting |
| KL340399 | KL340399 | Small-molecule activator | Undocumented | Sichuan Kelun Pharmaceutical Research Institute Co., Ltd. | Phase I | 2022.08.30 | Recruiting |
| NCT05514717 | XMT-2056 | ADC | Undocumented | Mersana Therapeutics | Phase I | 2023.01.24 | Recruiting |
| NCT04167137 | NCT04167137 | CDN | i.t. | i.t. | Phase I | 2019.11.01 | Unknown status |
Abbreviations: i.m.: intramuscular; i.t.: intratumoral; i.v.: intravenous; s.c.: subcutaneous; ADC: antibody-drug conjugate.
CDNs and structurally similar chemical compounds form the first-generation STING agonists prompted trends toward STING-based immunotherapy. However, the development of this type of CDN was stopped in 2021, because of unfavorable outcomes in phase II trials, in which the response rate was less than 10% [7]. The delivery efficacy of CDNs and related agonists is markedly limited [32]: even intratumoral injection of CDN has not shown satisfactory potency in clinical trials [33]. Naked agents, such as small-molecule STING agonists, have been found to be rapidly cleared and inefficiently delivered to tumor tissues [8, 9]. To date, these disadvantages have prevented STING-based immunotherapy from reaching the pharmaceutical market [34].
The first-generation STING agonists, derivatives of CDNs, exhibit fast clearance in vivo and poor membrane permeability, and require intratumoral administration, thus resulting in limited efficacy. Therefore, novel generation STING agonists are being developed to achieve systematic administration, increase the tumor distribution concentrations of compounds, and broaden the drug treatment window.
Because of the gap between the development and clinical application of first-generation STING agonists, efficacious delivery strategies and generation of novel STING agonists have attracted research attention [35]. Herein, we focus on the generation of novel STING agonists/potentiators and strategies to robustly promote immunotherapy.
4. DELIVERY STRATEGIES
4.1 Novel administration routes
The substantial role of STING in cancer immune surveillance has motivated leveraging of STING agonists as therapeutics to enhance antitumor immunity [36, 37]. Because of the high water solubility and polar surface area, the first-generation of STING agonists are typically administered through intratumoral injection. These STING agonists are quickly cleared from tumor sites and exhibit poor pharmacokinetic properties. However, repeated administration is not always feasible for the requirement of image-guide equipment and good patient compliance [35]. Ideal therapy scenarios with efficient delivery of therapeutics that bolster anti-tumor immune responses are needed [38].
Pulmonary administration delivers drugs directly to the lungs and has benefits of high drug concentrations in lung lesions, low systemic adverse effects, and ease of administration [39]. Jin et al. have developed an inhaled immunotherapeutic chitosan (CS)-anti-programmed death-ligand 1 (PDL1) complex for combatting lung cancer [40]. CS is a natural cationic copolymer that interacts with negatively charged sialic acid in mucins [41]. Moreover, CS engages the cGAS-STING pathway, thereby triggering the innate and adaptive immune systems [42]. Assembled with anti-PDL1, CS forms a nano complex that promotes the infiltration of immune cells around tumor lesions via repeated inhalation. Because of its efficient retention, transmucosal behavior, and adjuvant effect, CS has opened a new avenue for lung metastasis immunotherapy. Another inhalable nanoparticulate STING agonist, NP-cGAMP, has been reported by Liu et al. [43]. The STING agonist cGAMP has been loaded in phosphatidylserine-coated liposomes. Inhalation of NP-cGAMP has been found to lead to rapid distribution in mouse models of lung metastasis. Inhaled NP-cGAMP combined with fractionated radiation has been found to stimulate STING signaling and elicit systemic anticancer immunity.
Oral administration is another route that has favorable patient compliance, and avoids pain and contamination [44]. More than 65% of clinically applied drugs have been designed as oral formulations [45]. Pan et al. have identified a non-nucleotide STING agonist called MSA-2, which has been found to induce complete tumor regression in more than 80% of treated animals via oral administration [46]. Two MSA-2 molecules assemble and not only stack against Tyr167 from each STING subunit but also interact with surrounding side chains [47]. MSA-2 effectively inhibits tumor growth after oral administration of 20–200 mg/kg. Another non-nucleotide STING agonist, SR-301, has been identified by Chin et al. [48]. Because of its outstanding bioavailability (%F = 32.2), 15 mg/kg oral once-per-day dosing with SR-301 for 18 days has been found to decrease tumor burden in a B16F10 model.
Intravenous injection is the most widely used administration route for immunotherapy and is applicable for even metastatic tumors [49]. Generally, intravenous injection has been stymied by rapid degradation and insufficient tumor accumulation. Owing to a lack of understanding of pharmacokinetics, and a lack of evidence regarding efficacy and dosage, determining optimized administration procedures remains a challenge. With the advantages of prolonged circulation, targeted delivery, and/or stimuli response, novel nano-formulations and delivery strategies have been designed to activate the STING signaling pathway, reverse the immunosuppressive tumor microenvironment (TME), and improve immunotherapy ( Table 1 ).
4.2 Protection from degradation
To avoid unnecessary immune responses under normal cellular conditions, mammals have evolved to constitutively express deoxyribonucleases (DNase I, DNase II, and DNase III), which directly affect the length, concentration, and persistence of dsDNA, and negatively regulate cGAS activation [50, 51]. DNase I is a secretory protein present at approximately 3–10 ng/mL in blood and approximately 200 ng/mL in urine [52]. DNase I is expressed in most major organs, including the pancreas, kidneys, small intestine, and liver. Similarly, DNase II has been found in almost all human tissues and is preferentially located in lysosomes, but is also found in saliva, blood, urine, and testicular fluid [53]. The existence of DNase I and DNase II in the blood circulation and organs impedes the direct application of nucleic acid-like STING agonists via systemic administration. ENPP 1 poses another obstacle to STING-mediated immunotherapy. ENPP 1 hydrolyzes cGAMP and thus enables tumors to “cool” by decreasing STING activation [54]. Upregulated ENPP 1 has been observed in metastatic and chromosomally unstable tumors [54]. Efficient delivery of cGAMP remains a challenge because of its short half-life.
Though exogenous dsDNA finally enters the cytoplasm, human 3′ repair exonuclease 1 (TREX1) waited there. TREX1 degrades cytosolic nucleic acid and is a major regulator of cytosolic DNA metabolism [50, 55]. Notably, reactive oxygen species (ROS) triggered by ionizing radiation oxidize intracellular DNA bases, and the resultant steric hindrance partially inhibits TREX1-mediated degradation [56]. However, the IFN-stimulated gene TREX1 is upregulated during high doses of radiation and subsequently inhibits STING activation [57]. Repeated irradiation has been demonstrated to amplify IFN-β generation at dosages that do not induce TREX1 [58]. Comprehensive consideration of these inherent degradation mechanisms is critical in immunotherapy.
To protect agonists against these degradation mechanisms, researchers have used several approaches. The first approach modifies the chemical structures of CDNs. Lioux et al. have reported novel CDNs consisting of adenosine and inosine [59]. These novel CDNs contain one or two 2′-fluoro-2′-deoxyriboses and/or bis-phosphorothioate linkages. Compared with natural 2′, 3′-cGAMP, these modified structures endow the novel agonists with resistance to enzymatic cleavage and potent STING activation ability. In a parallel effort, Marketa et al. have found that masking the negative charges of CDNs with acyloxy methyl and isopropyloxycarbonyl phosphoester modifications improves their activity ( Figure 3B ) [60].
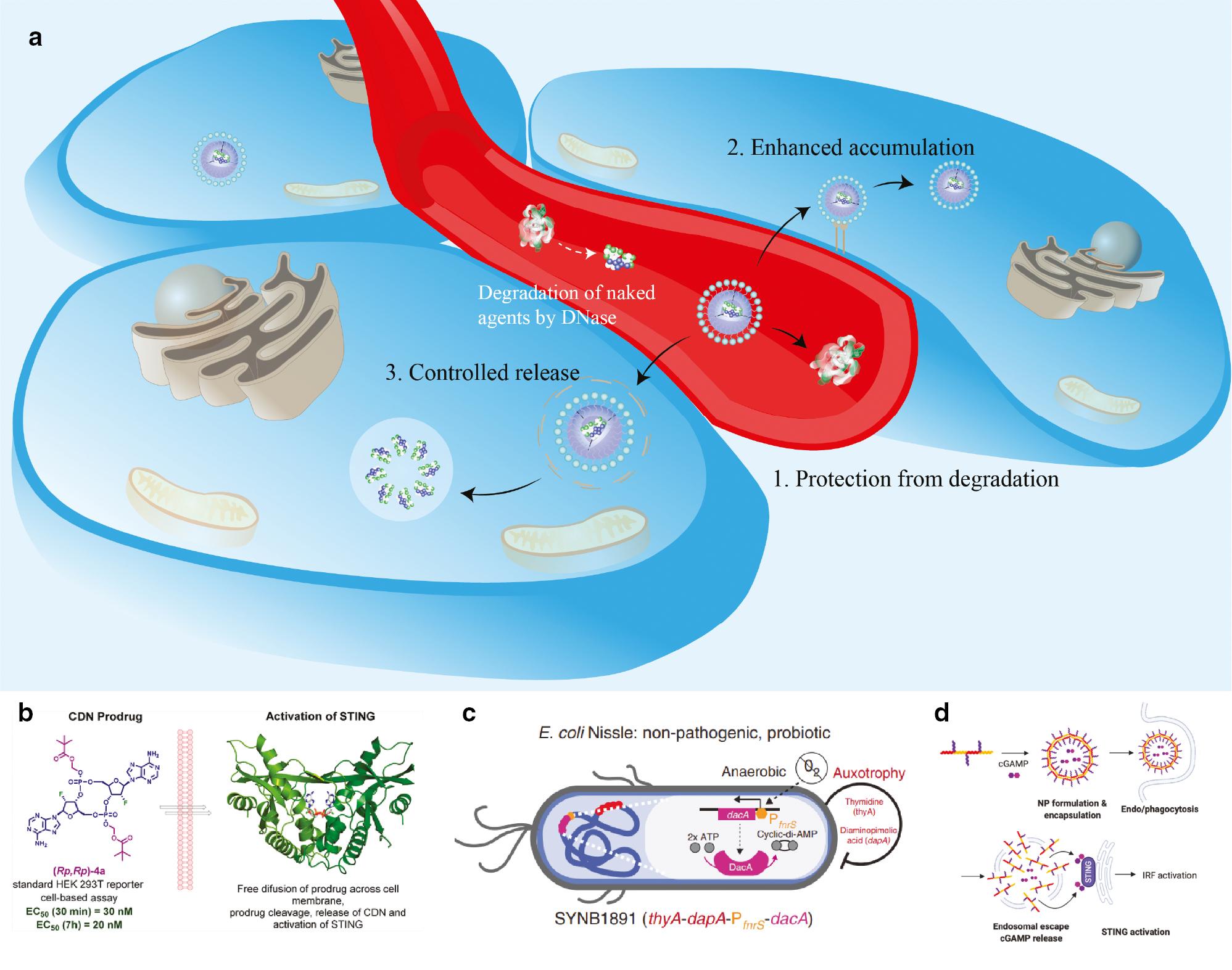
Nanotechnology-based strategies enhance STING agonist delivery to cancer cells.
a. The strategies include protection from degradation, enhanced accumulation, and controlled release. Various liposomes, polymers, and biomaterials have been used to cross barriers in STING activation. b. Phosphoester prodrugs of CDNs accelerate cellular uptake, and the prodrug moieties are cleaved rapidly [60]. Copyright © 2021, American Chemical Society. c. As a living biotherapeutic platform, modified E. coli produce high levels of cyclic di-AMP and induce potent type I IFN production [61]. Copyright © 2020, The Author(s). d. Copolymers with pH-responsive and membrane-destabilizing activity enhance intracellular delivery of CDN [62]. Copyright © 2020, Wiley-VCH GmbH.
Another method involves protecting agonists with undegradable umbrellas ( Figure 3 ). Shae et al. have designed cGAMP-encapsulated polymersomes (PEG2kDa-DBP5kDa vesicles) denoted STING-NPs [63]. The nanoparticles increase the biological potency of cGAMP and enhance STING signaling in the TME and sentinel lymph nodes. Through an endosomal escape mechanism, STING-NPs enhance the cytosolic delivery of cGAMP and preferentially activate STING in myeloid cell populations. This rational design has been found to elicit complete and robust antitumor immunity in tumor re-challenge. Another endosomolytic polymer, D-PDB, has been identified and used by Garland et al. to encapsulate phosphorothioate-capped 95-BP dsDNA [64]. This nanoparticle formulation confers deoxyribonuclease resistance and potent activation of the STING pathway. Liposomes, another widely used carrier, have also been used to encapsulate cGAMP and other nucleic-acid-based adjuvants [65]. These well-established drug delivery systems have enabled the stability of agonists and enhanced cellular activity after internalization.
4.3 Enhanced accumulation
Generally, CDN molecules are anionic and pose difficulties in cytosolic delivery. David et al. have used cationic polymers to deliver CDNs inducing a robust immune response; this method has traditionally been used to deliver DNA and small interfering ribonucleic acids [66]. Similarly, Koshy et al. have reported that liposomal cGAMP increases cell association and uptake to a greater extent than the free form [67]. Liposomal encapsulation increases pro-inflammatory-gene induction and anti-tumor activity in metastatic tumors. Regression of treated tumors and the establishment of immunological memory have indicated improvements in the activity and potential clinical utility of STING agonists. Thus, delivery strategies for nucleic acids may facilitate STING-based immunotherapy.
Given the high heterogeneity of STING-associated genes, concerns persist regarding whether all human populations might be responsive to direct cGAMP administration [68]. In complex with cGAMP, a self-assembled tetrameric structure of the transmembrane-domain-deficient STING protein has been found to effectively trigger STING signaling [69]. Thus, researchers have developed a cGAMP delivery strategy using a recombinant, transmembrane-deficient STING as a carrier in the form of a ribonucleoprotein complex. Even in STING-deficient cell lines, this approach promotes a strong humoral response and antigen-specific T cell activation, thus providing a new paradigm for vaccinology and immunotherapy.
For various bioactivities and specific biodistributions, a series of biomaterials have been used as immunotherapeutic carriers. Exosomes are natural extracellular vesicles with vast potential for drug delivery [70–73]. Because of their natural ability to be taken up by cells and their excellent biocompatibility, exosomes have been engineered to deliver cGAMP in a study by Kathleen et al. [74]. Exosomes display superior uptake by DCs and lead to accumulation of activated CD8+ T cells in B16F10 tumors. Kauke and colleagues have developed anti-Clec9A exosomes targeting conventional type I DCs [75]. Loaded with CDNs, anti-Clec9A exosomes achieve tumor control by delivering a single agent. Compared with controls, engineered exosomes have been found to induce immune responses against tumor-associated antigens. Another targeted living biotherapeutic platform has been reported by Daniel et al., who have engineered non-pathogenic E. coli to produce cyclic di-AMP and contact with antigen-presenting cells (APC) in tumors ( Figure 3C ) [61]. This synthetic biological method includes dual safety and biocontainment features, and activates complementary innate immune pathways.
4.4 Controlled release
Drug release triggered by specific stimuli has improved therapeutic outcomes ( Table 2 ). Responsive pH-triggered delivery is one of the most prevalent approaches to targeting the acidic extracellular TME and intracellular organelles [82]. Miyabe et al. have induced IFN-β in Raw264.7 cells with pH-sensitive c-di-GMP/YSK05 liposomes [76]. C-di-GMP liposomes induce significantly higher expression level of CD80, CD86, and MHC class I, and improve the innate immune system response. Kawai et al. have reported plasmid-DNA-encapsulating lipid nanoparticles with SS-cleavable and pH-activating properties [77]. Dual sensing motifs respond to the intracellular environments and exhibit extensive therapeutic antitumor effects. Stimulus-responsive polymers have also been described. Through increased intracellular CDN uptake and endosomal destabilization, Nguyen et al. have established an efficient STING-based immune response activation strategy ( Figure 3D ) [62]. Li et al. have developed a pH-sensitive polymer with a seven-membered ring associated with a tertiary amine (PC7A) [83]. In contrast to other CDNs, PC7A is bound to a non-competitive STING site distinct from the cGAMP pocket, and it stimulates prolonged production of pro-inflammatory cytokines. Even in cGAMP-resistant genetic variants, the combination of PC7A and cGAMP leads to therapeutic outcomes in mice bearing various types of tumors.
Delivery strategies for nanotechnology-based STING agonists.
| Nanomedicine | Formulation | Payload | Enhanced accumulation | Stimulus sensitivity | Tumor model | Reference |
|---|---|---|---|---|---|---|
| Liposomal cGAMP | Liposome | cGAMP | In APCs | - | In situ and lung metastatic | [67] |
| C-di-GMP/YSK05 | Liposome | Cyclic di-GMP | In macrophages | pH | Lymphoma | [76] |
| ssPalmE-LNP | Liposome | Plasmid DNA | In macrophages | pH | Lymphoma and melanoma | [77] |
| CDN-PBAE | CDNs | In APCs | - | Melanoma | [66] | |
| PEGMA-co-DEAEMA-co-BMA copolymer | Polymer | CDNs | In TME | pH | Melanoma | [62] |
| STING NPs | Polymer | cGAMP | In TME | - | Melanoma and breast adenocarcinoma | [78] |
| STINGel | Hydrogel | CDNs | In TME | - | Head and neck cancer | [79] |
| HNVs | Hybrid cell-membrane nanovesicle | SIRPα variants and cGAMP | In macrophages | - | Melanoma | [80] |
| Sting-NPs | Polymer | cGAMP | In APCs | - | Human melanoma | [81] |
SIRPα: signal regulatory protein alpha; TME: tumor microenvironment; CDC1: conventional type I dendritic cell.
Particles formulated with the biodegradable polymer acetylated dextran have been used to enhance intracellular delivery by the Watkins group [84]. These polymers resulted in tumor growth inhibition at 100–1000-fold-lower doses than published doses of soluble cGAMP. Analysis of immune cell depletion has confirmed that NK cells and T cells are responsible for the anti-tumor function. Similarly, biodegradable poly (beta-amino ester) (PBAE) nanoparticles contribute to a robust immune response at >100-fold-lower extracellular CDN concentrations than free CDN [66]. Recently, Wehbe et al. have analyzed the pharmacokinetic and pharmacodynamic properties of polymer-based nanoparticles [78]. Nanoencapsulated STING NPs have been found to prolong the half-life of encapsulated cGAMP by 40-fold. Intravenous administration of STING NPs reprograms the TME, thus eliciting a >20-fold increase in the influx of CD4+ and CD8+ T cells. Monotherapy with STING NPs has been found to achieve a >50% and >80% decrease in melanoma and breast adenocarcinoma models, respectively. Strong innate immune responses are induced by pathways activated by polymer-based STING administration.
Other formulations for delivery of STING agonists have been developed to achieve sustained release. The biocompatibility, design flexibility, high swelling ratio, and hydrophilic nature of hydrogels make them suitable for immunomodulation [85]. An injectable peptide hydrogel called STINGel for controlled release of CDN has been reported by Leach et al. [79]. This positively charged hydrogel achieves an 8-fold slower release rate than that of standard collagen hydrogel. In a model of head and neck cancer, STINGel markedly prolongs overall survival. Toward the same goal, Lu et al. have developed microfabricated polylactic-co-glycolic acid particles [86], which remain at the resection site and release encapsulated STING agonist through a programmable pulse at predetermined time points. This strategy mimics multiple injections, thereby preventing recurrence after surgery, and inhibits the growth of tumors that are difficult to reach.
5. ACTIVATION STRATEGIES
5.1 Novel agonists
With advanced chemical synthesis and high-throughput screening technology, substantial numbers of novel agonists have been identified and constructed in recent years. The new structures have enabled new administration routes or resistance to degradation, and other impressive capabilities.
Given that the responses of the above-mentioned STING variants to CDNs are variable, a pan-genotypic STING agonist is needed. In innate immunity activation, CDNs use a U-shape conformation with two nucleobases in a π-parallel position after binding STING protein dimers [87]. To rigidify the CDN in the binding conformation, Kim et al. have synthetized a novel macrocycle-bridged agonist E7766 [88]. By locking the STING active CDN conformation, E7766 displays superior and pan-genotypic activity against the major human STING variants to cGAMP.
Miao et al. have identified ionizable lipid-like materials that stimulate the STING pathway and have developed a combinatorial library [89]. The common structures of these lipids are unsaturated lipid tails, dihydromidazole linkers, and cyclic amine head groups. Ultimately, the authors synthesized and identified optimized formulations inducing a robust immune response and inhibiting tumor growth. Moreover, these lipids can serve as carriers for mRNA delivery.
Metalloimmunotherapy involves immune regulation. Mn2+ increases the binding affinity of cGAS toward cytosolic DNA [90]. Thus, Mn2+-coordinated CDN nanoparticles have been developed [91, 92]. Elevated cytokine production, owing to the synergistic effects of Mn2+ and CDNs, leads to T cell immunity activation. Recently, Zn2+-based nanoclusters have been demonstrated to potentiate the activation of STING [93]. Meanwhile, because other metals, such as Co2+, exhibit STING activation capability, metalloimmunotherapy may shift the current understanding of cancer treatment [94].
5.2 Antigen-presenting-cell activation
APCs, including macrophages and DCs, engulf living or dead tumor cells, and exhibit tumor-associated antigens through cross-presentation [95]. However, tumor cells demonstrate low immunogenicity and enable escape from recognition of APCs and avoidance of subsequent immune responses [5]. Robert et al. have evaluated the activity of cGAMP encapsulated in acid-sensitive acetylated dextran polymeric nanoparticles [96]. These nanoparticles passively target APCs for intracellular release. The polymeric nanoparticles enhance type I interferon responses nearly 50-fold in vivo, and expand germinal center B cells and memory T cells, thus providing a potent adjuvant for humoral and cellular immunity.
Cytotoxic cationic silica nanoparticles induce acute necrotic tumor cell death and release tumor-associated antigens. By complexing with c-di-GMP, these nanoparticles activate tumor-infiltrating APCs and markedly increase the expansion of antigen-specific CD8+ T cells [97]. Additionally, APC activation is promoted by protein X (Prx), a transmembrane glycoprotein that naturally occurs on exosomes. Thus, exosomes have been engineered by Jang et al. and loaded with a STING agonist (exoSTING) that selectively activates STING in APCs [98]. This strategy increases the tumor-specific T cell response and eliminates non-injected abscopal tumors.
Moreover, tumor cells overexpress the “don’t eat me” signaling molecular CD47 and evade subsequent spontaneous phagocytosis [99]. CD47 binds the phagocytosis checkpoint protein signal regulatory protein α (SIRPα) on tumor-infiltrating APCs and hinders tumor elimination. To target this phagocytosis checkpoint, Lu et al. have co-delivered a small interfering RNA and cGAMP into APCs via polymeric nanoparticles (NPsiSIRPα/cGAMP) [100]. siSIRPα mediates SIRPα silencing and facilitates APC capture of tumor antigens. Subsequent STING activation enhances the function of APCs and induces systemic anti-tumor immune responses by reversing the immunosuppressive phenotype of APCs.
5.3 Drainage to lymph nodes
Cancer vaccines establish long-term immune memory and inhibit tumor progression, recurrence, and metastasis [101] ( Table 2 ). By delivering tumor antigens to APCs, cancer vaccines enable activation of tumor-specific immune responses [102]. As modulators of the innate immune system, STING agonists have been tested as an adjuvant to increase vaccine immunogenicity and efficacy [103, 104] ( Table 3 ). STING-activating nano-vaccines have advantages over in situ nanotechnology-based immunotherapy, thus paving the way to multilevel anti-tumor strategies [33, 107]. Hanson et al. have encapsulated CDNs with PEGylated lipid nanoparticles to form the nano-vaccine NP-cdGMP [108], which increases CD8+ T cells and enhances therapeutic effects by redirecting this nano-vaccine to draining lymph nodes. Combined with a liposomal peptide antigen, NP-cdGMP robustly induces tumor-associated CD4+ T cell expansion and germinal center B cell differentiation. Recently, high-Z sensitive radiotherapy has been reported to trigger abundant antigen and adjuvant release [109]. This strategy successfully primes robust CD8+ T cell-dependent immunity and potentiates checkpoint-blockade immunotherapies. Thus, the combination of a high-Z sensitizer and radiotherapy-mediated STING activator facilitates DC activation and enhances immunotherapy.
STING-mediated nano-vaccines for cancer treatment.
| Nano vaccine | Formulation | Payload | Tumor model | Enhanced ICBs | Reference |
|---|---|---|---|---|---|
| NanoSTING-vax | Polymer | cGAMP and peptides | Melanoma and colon cancer | + | [63] |
| Co-encapsulated CpG ODN + cGAMP | Liposome | OVA, TLR9 and STING ligands | Melanoma | - | [65] |
| cGAMP-STING△TM | Ribonucleoprotein complex | cGAMP | Melanoma and colon cancer | - | [69] |
| Mn-cGAMP | Nanoparticle | Mn2+ and cGAMP | Melanoma | + | [91] |
| Acid-activatable nanovaccine | Nanoparticle | Neoantigen and DMXAA | Melanoma and breast cancer | + | [105] |
| PC7A-NP | Mixture of antigen and polymeric nanoparticle | OVA and polymer NPs | Melanoma, colon cancer and human papilloma virus E6/E7 | + | [106] |
| STING–PC7A | Polymer | PC7A and cGAMP | Colon cancer and human sentinel lymph nodes | - | [83] |
OVA: ovalbumin; TLR9: Toll-like receptor 98.
Selective triggering by the TME facilitates on-demand release into APCs in lymph nodes. Kocabas et al. have co-encapsulated STING ligands with Toll like receptor 9 (TLR9, a member of the Toll-like receptor family that uses pathogen-associated molecular patterns as ligands to induce the innate immune response) and a model antigen ovalbumin within pH-sensitive liposomes [65]. This delivery system has achieved a heightened Th-1 response. Pronounced protective vaccine efficacy has been achieved by loading with dual ligands. Increased CD8+ T cells and decreased M2 macrophages in the tumor bed induce reversal of the immunosuppressive TME. NanoSTING-vax is a cancer nanoplatform co-delivering STING agonists and peptide antigens [63]. This pH-sensitive endosomolytic polymer enhances CD8+ T cell responses by eliciting cytokine production, co-stimulatory marker expression, and antigen cross-presentation. Similarly, Zhou et al. have reported an acid-responsive polymeric nano-vaccine fabricated by integrating the STING agonist DMXAA and a neoantigen [105]. Stimulus-triggered designs promote nano-vaccine accumulation at lymph nodes and neoantigen release into the cytoplasm.
5.4 Remodeling of the immunosuppressive microenvironment
In the TME, most macrophages display an M2-like functional profile and induce an immunosuppressive microenvironment [110]. Therefore, macrophage reprogramming can be leveraged to augment immunotherapy. Ning et al. have shown that liposomal STING-directed macrophages are reprogrammed from a pro-tumor M2 phenotype to an anti-tumor M1 phenotype, thus increasing IFN-γ-producing T cells [111]. This strategy modulates the TME and effectively controls PD-L1-insensitive tumors.
Several types of cancers poorly respond to immune checkpoint blockers (ICBs), owing to their immunosuppressive TMEs. Switching from a “cold” tumor to a “hot” tumor via STING activation is believed to enhance the therapeutic effects of ICBs [112]. Luo et al. have reported a minimalist nano-vaccine consisting of an antigen and PC7A NP. The combination of the PC7A nano-vaccine and an anti-programmed death-1 antibody (αPD-1) exhibits notable synergy, thus resulting in 100% survival in tumor-bearing mice over 60 days. Re-challenge of these tumor-free animals has been found to result in in complete inhibition of tumor growth, thus suggesting a robust strategy for boosting immunological memory. Most STING-based nano-vaccines combined with ICBs have demonstrated enhanced antitumor efficacy [63, 66, 78, 91, 105]. Overcoming intratumoral immune tolerance promotes the responses of certain tumors to immunotherapy.
The tumor vasculature is usually structurally malformed and functionally anergic, thus impeding T cell infiltration and effector functions [113, 114]. Vascular normalization may alleviate TME immunosuppression and enhance immunotherapy. Yang et al. have combined STING agonists with vascular endothelial growth factor receptor 2 (VEGFR2) blockade and ICB (αPD-1 or anti-cytotoxic T lymphocyte antigen 4 antibody (αCTLA-4)) [115]. This immunotherapy is highly effective, thus leading to complete regression of immunotherapy-resistant tumors. Alleviation of vascular abnormalities and TME immunosuppression provides a rationale for combined immunotherapy.
6. APPLICATION STRATEGIES
6.1 Surgery
Surgery remains the major treatment for resectable tumors. However, surgery not only eradicates tumor tissues but also removes tumor-specific antigens and immune cells [116]. Subsequently, an immunosuppressive microenvironment forms when the surgical wound heals, thus potentially promoting tumor recurrence and metastasis [117] ( Figure 4A ). To prevent local tumor recurrence, Chun et al. have placed biodegradable hydrogels with extended release of STING agonists in tumor resection sites [121], thus resulting in activation of innate and adaptive immune responses; prevention of local tumor recurrence; eradication of existing metastases; and further reversal of the post-resection immunosuppressive microenvironment to an immunostimulatory one.
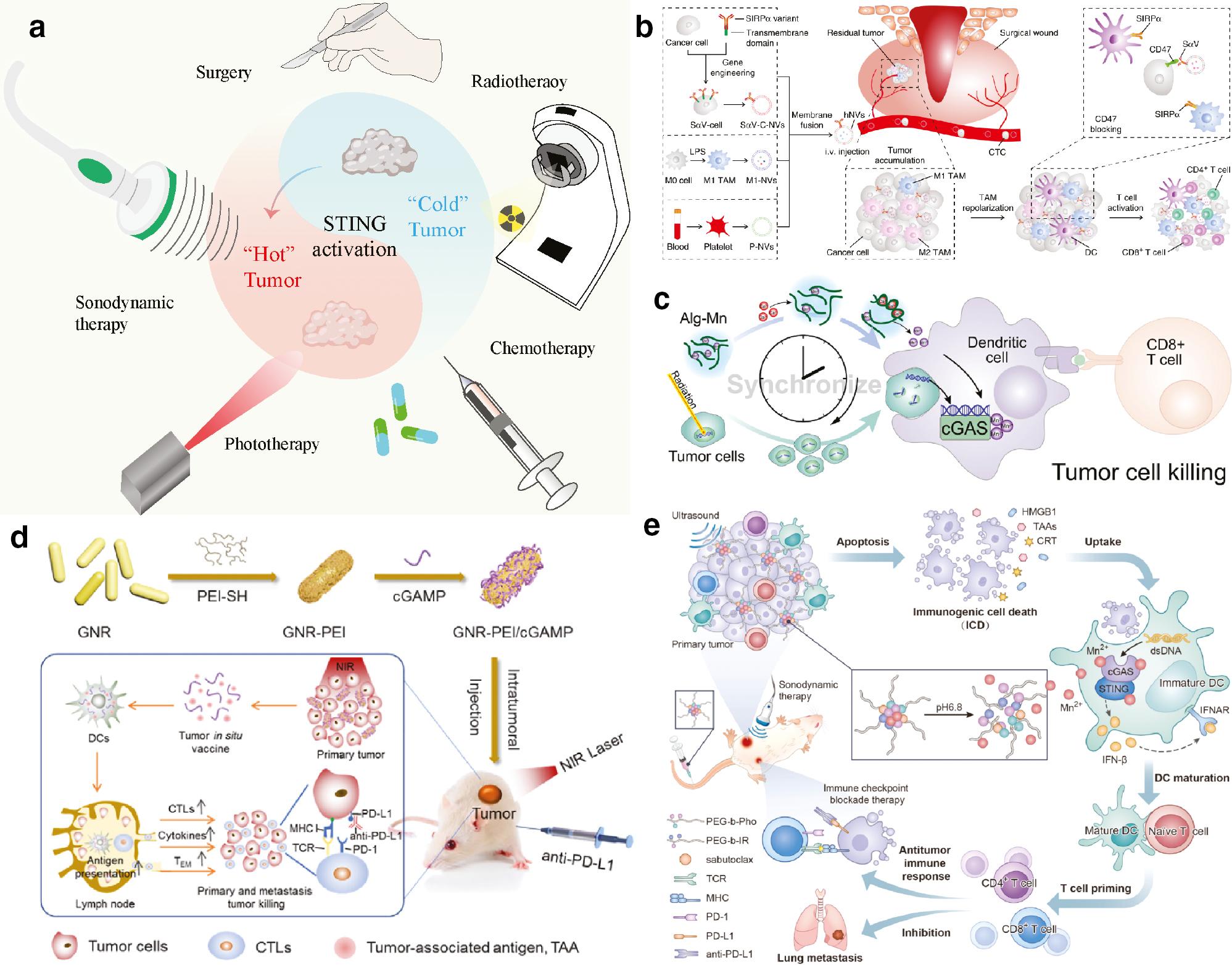
Application strategies in STING-mediated cancer treatments.
a. Until recently, traditional anti-cancer therapies (e.g., surgery, radiotherapy, and chemotherapy) served as major strategies. The success of cancer eradication is increasingly being understood to rely on the induction of a continuous immune response, which converts “cold” to “hot” tumors. With nanotechnology, STING-mediated traditional treatments (surgery, radiotherapy, and chemotherapy) and novel therapeutic interventions (e.g., phototherapy and sonodynamic therapy) have been developed to improve anti-cancer efficacy. b. In a breast cancer postoperative model, hybrid cell membrane nanovesicles lead to potent tumor inhibition by loading cGAMP [80]. Copyright © 2020, This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply. c. Maintaining Mn2+ in tumor in accordance with radiotherapy induced DNA accumulated in the cytosol helps to induce cGAS-STING pathway activation and promote radiotherapy-induced [118]. Copyright © 2021 Elsevier B.V. All rights reserved. d. In complex with cGAMP, positively charged polyethyleimine-modified gold nanorods serve as an in situ tumor vaccine under near-infrared laser-triggered phototherapy [119]. © 2020 The Authors. Published by Elsevier Ltd. e. SDT-mediated ICD effects and Mn2+ promote activation of the cGAS-STING pathway, thus efficiently inhibiting the growth of distant tumors [120]. © 2022 Elsevier Ltd. All rights reserved.
Engineered nanovesicles coated with membrane structures similar to those of their parent cells have also attracted attention [122]. Rao et al. have reported that hybrid cell membrane vesicles display signal regulatory protein alpha (SIRPα) variants ( Figure 4B ) [80]. SIRPα on macrophages interacts with CD47 on cancer cells. The “don’t eat me” signal expressed by cancer cells polarizes tumor-associated macrophages from M2 to M1. Loading of STING agonists in hybrid cell membrane vesicles has been found to lead to potent tumor inhibition.
6.2 Radiotherapy
Radiotherapy is a universally available and effective treatment that can be precisely tailored to tumor sites. Research has indicated that radiotherapy enables the introduction of effective anti-cancer immune responses. However, systemic anti-cancer immune response is rarely observed in clinical practice. According to Nature Reviews Clinical Oncology, combining radiotherapy with immunotherapy helps induce a long-lasting immune response [123]. As early as the 1990s, researchers observed the enhancement of radiotherapy with DMXAA [124–126]. Radiotherapy induces DNA double-strand breaks [127]. Free DNA in the cytoplasm bound by cGAS activates the STING pathway, thus enabling synergistic control of local and distant tumors with STING agonists ( Table 4 ).
Application strategies for nanotechnology-based STING interventions.
| Application strategy | Nanomedicine | Payload | Tumor model | Reference |
|---|---|---|---|---|
| Radiotherapy | NP-cGAMP | cGAMP | Metastatic breast cancer | [43] |
| Surgery | STING-RR | cGAMP | Breast cancer | [121] |
| Surgery | Hybrid cell membrane nanovesicles | cGAMP | Breast cancer and melanoma | [80] |
| Radiotherapy | Hafnium oxide nanoparticles | Hafnium oxide nanoparticles | Colorectal cancer | [128] |
| Radiotherapy | Alg-Mn | Mn2+ | Colon cancer and melanoma | [118] |
| Chemotherapy | SN38-NPs | SN38 | Breast cancer | [129] |
| Chemotherapy | PS3D1@DMXAA | DMXAA and SN38 | Breast cancer | [130] |
| Chemotherapy | Mn3O4@Au-dsDNA/DOX | Au-dsDNA and DOX | Breast cancer | [49] |
| Chemotherapy | Supramolecular hydrogel | CDNs and camptothecin | Breast cancer | [131] |
| Phototherapy | GNR-PEI/cGAMP | Polyethyleimine-modified gold nanorods and cGAMP | Osteosarcoma | [119] |
| Phototherapy | Melanin-loaded DNA adjuvant hydrogel | C-di-GMP, CpG, and melanin | Colon cancer and breast cancer | [132] |
| Sonodynamic therapy | PIMS-NPs | Mn2+ | Breast cancer | [120] |
| Radiotherapy | PC7A nano-vaccine | Antigen and PC7A | Metastatic lung cancer | [133] |
Because of radiation dose limitations, existing radiotherapy cannot induce sufficient ROS generation to trigger the leakage of broken dsDNA into the cytosol. Marill et al. have reported that hafnium oxide nanoparticles can be activated by radiotherapy [128]. Compared with radiotherapy alone, use of hafnium oxide nanoparticles activated by radiation generates significantly more double-strand breaks and micronuclei; consequently, greater STING pathway activation can be obtained with higher levels of radiotherapy. These results have suggested potential applications of this nanotechnology-based strategy in radiation reduction therapy. A phase I/II trial has been launched to administer hafnium oxide nanoparticles, radiotherapy, and PD-1 inhibitors in patients with advanced cancers (NCT03589339).
After radiotherapy, some time is required for dsDNA to leak into the cytoplasm. Therefore, the release of Mn2+ in tumors should be consistent with the accumulation of dsDNA [118, 134]. After radiotherapy, we have found that sustained release of Mn2+ by alginate nanoparticles synchronizes with the time-dependent accumulation of DNA damage and synergistically amplifies STING signaling activation ( Figure 4C ) [118]. The metabolic process of Mn2+ can also be monitored in real time through MRI imaging. Greater than 90% inhibition of tumors and prolonged survival times were observed in that study, thus promoting radiotherapy-induced immunity. By activating STING pathway signaling, metalloimmunotherapy robustly enables triggering of a radiotherapy-induced immune response.
6.3 Chemotherapy
Chemotherapeutic drugs, such as etoposide and camptothecin, promote DNA damage, thus leading to the release of DNA fragments into the cytosol and STING pathway signaling activation [135]. Moreover, some chemotherapeutic agents, such as cisplatin, show myelosuppressive effects and are likely to induce immunosuppressive status by killing of circulating lymphocytes [136]. STING activation compensates for the negative effects of chemotherapy on immune cells by activating the immune response through an alternative pathway [137].
By synergy between chemotherapeutic agents and STING agonists, nanotechnology potentiates systematic anti-cancer effects in chemoimmunotherapy. Zhao et al. have presented an approach for activation of STING signaling via nanoplatform-delivered DNA-targeting chemoagents [129]. The compound 7-ethyl-10-hydroxycamptothecin (SN38) has been identified as the most potent chemotherapeutic agent for stimulating interferon-β secretion via STING activation. SN-38 nanoparticles suppress 82.6% of tumor growth and consequently have inspired DNA-targeting drugs in nano-formulations for chemoimmunotherapy. Liang et al. have described a combined strategy concurrently delivering SN38 and DMXAA to tumors by using copolymer nanoparticles denoted PS3D1@DMXAA [130]. PS3D1@DMXAA enhances antigen cross-presentation and induces the conversion from an immunosuppressive to an immunogenic TME. The synergistic function between SN38 and STING activation has shown potent therapeutic efficacy and elicited remarkable therapeutic benefits in combination with ICBs.
Zhou et al. have designed a nano-formulation of Mn3O4@Au-dsDNA/doxorubicin (DOX) to address the challenge of immunosuppressive tumors [49], in which dsDNA was chosen to activate STING for immunotherapy, and DOX was used as a model drug for chemotherapy. The authors have shown that anti-tumor immunity, combined with innovative nanotechnology, chemotherapy, and targeted therapy, suppresses tumor growth and prolongs survival rates in vivo, thus facilitating cancer treatment. A self-assembled supramolecular hydrogel constructed from CDNs and the peptide-camptothecin conjugate has been reported by Wang’s team [131]. This strategy has led to long-term immunological memory and systemic immune surveillance, thus protecting mice against tumor recurrence and the formation of metastases.
6.4 Phototherapy
Phototherapy modalities include photodynamic therapy (PDT) and photothermal therapy [138]. Under laser irradiation, PDT induces ROS-mediated tumor damage [139], whereas photothermal therapy increases temperatures at tumor sites [140]. This precise and minimally invasive approach induces necrosis of tumor cells, which may release intracellular constituents. By releasing tumor-associated antigens and dsDNA, phototherapy enables STING activation and induces an anti-tumor immune response. On the basis of this mechanism, some researchers have explored combined strategies of phototherapy and immunotherapy. Zhao et al. have reported that positively charged polyethyleimine-modified gold nanorods (GNR-PEI) can be complexed with cGAMP through electrostatic interaction, thus forming GNR-PEI/cGAMP ( Figure 4D ) [119]. Near-infrared-laser-triggered photothermal therapy for primary tumors has been used to generate an in situ tumor vaccine, which recruits DCs to capture tumor-associated antigens. cGAMP stimulates the expansion and activation of DCs, and the specific immune-killing effects of CTLs. The combination of this localized tumor vaccine and ICBs reverses the immunosuppressive TME and increases killing of remaining and metastatic tumor cells.
Wu et al. have designed a novel melanin-loaded DNA adjuvant hydrogel, in which c-di-GMP serves as a STING agonist, and CpG serves as a TLR9 agonist [132]. Co-delivery of adjuvants (c-di-GMP and CpG) and photo-responsive melanin activates DC in tumor-associated draining lymph nodes and enhances the generation of CTLs. This biodegradable and biocompatible hydrogel formulation destroys primary tumors and activates a potent immune response to prevent re-challenge.
6.5 Sonodynamic therapy
Sonodynamic therapy (SDT) is a recently developed intervention with deep penetration and a non-invasive nature [141]. The interactions between ultrasound and tissue include absorption and cavitation effects, but not radioactivity damage [142]. Typically, the combination of ultrasound and sonosensitizer triggers a specific sonochemical reaction that generates ROS with extensive toxicity [143]. The synergy of the SDT-mediated immunogenic cell death (ICD) effect and STING activation is believed to boost anti-tumor immunity.
Dai et al. have integrated sonosensitizers (IR780) and Mn2+ into amphiphilic phenolic polymers and developed a nano-adjuvant ( Figure 4E ) [120]. The SDT-mediated ICD effect releases tumor-associated antigens, whereas Mn2+ increases the sensitivity of tumor-derived DNA in DCs by facilitating STING activation. This combinational action enhances both innate and adaptive immune responses, and thus is a promising candidate for combination therapy.
7. CONCLUSIONS AND FUTURE PROSPECTS
With further insight into the tumor immune environment and the rapid development of material science, new generations of STING-mediated anti-tumor strategies have been developed. Novel structures and various nano-formulations for activating STING signaling have induced a shift from immunosuppressive “cold” tumors to “hot” tumors. Novel administration routes and delivery strategies have improved responses to STING agonists. Moreover, efforts have focused on establishing STING-induced anti-cancer immunity. These new therapeutics, combined with traditional therapeutics. may be an emerging direction for reprograming the TME and ultimately achieving favorable therapeutic performance in clinical settings.
Notably, several issues remain to be addressed to achieve clinical translation. Drug leakage during circulation may lead to off-target toxic effects. Greater attention should be paid to the conjugation interaction effects of nanomaterials and STING agonists. To overcome biological barriers, the characteristics associated with transmembrane transport and intracellular delivery should be carefully considered. Molecular mechanisms such as reprogramming of the tumor immunosuppressive microenvironment and tumor antigen presentation await further exploration [144].
Immunotherapy via STING pathway modulation relies on the human immune system, thus potentially leading to significant differences among preclinical and clinical trials. Whether direct cGAMP administration enables responses in patients remains under debate [68]. Furthermore, plasma proteins that spontaneously sorb onto the surfaces of nanomedicines in the blood circulation may block the targeting agent and contribute to an unfavorable immune response, thus affecting the targeting yield of nano-formulations [145]. To apply preclinical strategies in clinical practice, studies must be conducted to gain a deeper understating of the interactions between STING-modulating nanomedicines and the host immune system in vivo.
Recent advances in nanotechnology-based anti-cancer strategies via the STING pathway were highlighted in this review. Novel-generation STING-modulating strategies are expected to facilitate optimized therapeutics designed for clinical applications. Furthermore, a deeper understanding of the TME and immune mechanisms may promote the design of new strategies, thereby accelerating anti-cancer immunotherapy development. STING-mediated strategies hold great promise for immune modulation and are not limited to cancer treatment.