
1. INTRODUCTION
Platelets are small anucleate subcellular fragments derived from the cytoplasm of bone marrow megakaryocytes (MKs) [1]. Approximately 200 billion platelets are produced every day by an average adult person at rest [2]. After their release from the bone marrow MKs into the bloodstream, platelets remain in the blood circulation for approximately 8–10 days. Human platelets are 1–3 μm in diameter and exhibit a plate-like discoid shape, which maximizes planar-surface interactions [3]. The platelet outer membrane consists of glycoproteins and has abundant integrins that facilitate adhesion and aggregation [4]. Platelets contain messenger RNA (mRNA) but no DNA, and thus can synthesize only a limited number of proteins. Platelets have multiple functional organelles (including endoplasmic reticulum, Golgi apparatus, and mitochondria) and three types of functionally relevant granules (α-granules, dense granules, and lysosomal granules) [5–7]. Each platelet contains approximately 50–80 α-granules, which in turn contain various proteins including membrane-bound adhesion receptors such as glycoprotein (GP) VI, GPIIb/IIIa, or the Willebrand factor (vWF) receptor GPIb-IX-V complex. These granules also contain vWF, fibrinogen, fibronectin, and vitronectin, as well as growth factors (e.g., insulin-like growth factor (IGF), transforming growth factor-beta (TGF-β), and platelet-derived growth factor (PDGF)), and numerous immunomodulatory molecules and chemokines (e.g., CCL3/MIP-1α, CCL5/RANTES, and CXCL4/PF4) [6]. Dense granules contain small non-protein molecules important for hemostasis and innate immunity, such as ATP, histamine, ADP, calcium, glutamate, and serotonin [7]. Lysosomal granules contain glycosidases, acid proteases, and cationic proteins, which have bactericidal activity [8].
Platelets are crucial mediators of hemostasis and thrombosis, and their basic functions are to maintain the integrity of the blood vessels and participate in the coagulation cascade at sites of vascular injury. After vascular damage, circulating platelets initially adhere to the subendothelial extracellular matrix (ECM) through a variety of receptors, including the collagen receptors α2β1 and GPVI, and GPIb-IX-V. Platelets, after firmly adhering to the subendothelial ECM, undergo diffusion, activation, and eventual aggregation and thrombus formation [9]. Activated platelets release granules and signaling molecules, which recruit and activate nearby platelets as well as other blood cells, such as erythrocytes and leukocytes, thereby reinforcing thrombus formation and preventing further bleeding [10]. Activated platelets are eventually cleared by hepatocytes and/or local phagocytes, such as macrophages and neutrophils, and activation of GPIb-IX signaling is a key trigger of this process [11–13]. Beyond their primary roles in hemostasis and thrombosis, platelets have roles in modulating inflammatory reactions and immune responses [14]. Platelets participate in immune responses through direct interactions with other immune cells, or by secreting immunomodulatory and polarizing molecules. Activated platelets have high surface expression of the adhesive molecule P-selectin, which has important roles in platelet interactions with immune cells expressing P-selectin glycoprotein ligand-1 (PSGL-1), such as lymphocytes, neutrophils, and monocytes. Platelets are involved in the direct activation of B and T lymphocytes, polynuclear neutrophils (PNNs) and dendritic cells via the CD40L/CD40 complex [15].
Recent studies have shown that tumors actively recruit platelets, thereby promoting cancer survival, progression, and metastasis. These findings have inspired the concept of using platelets as live carriers for tumor-targeted drug delivery. Platelets are also active constituents of the tumor microenvironment (TME) that coordinate and regulate the functions of various tumor-associated immune cells. Thus, interest is increasing in the use of tumor-associated platelets as targets for therapeutic modulation of the TME and augmenting the anti-tumor immune response. In this review, we present and discuss recent advances in tumor-associated platelets and their interactions with the TME. Special emphasis is placed on exploiting platelets for both antitumor drug delivery and immune modulation of the TME.
2. PLATELETS AND TUMORS
2.1 Thrombocytosis and cancer
Clinical studies have shown that patients with late-stage cancer frequently develop thrombotic complications [16, 17]. High platelet counts have been associated with poor prognosis and survival in many solid cancers, including lung, breast, kidney, glioblastoma, pancreatic, ovarian, and gastrointestinal cancers [18]. Several molecular mechanisms have been suggested to underlie the progression of cancer-associated thrombocytosis, which involves various tumor-associated cytokines and humoral factors directly or indirectly affecting megakaryopoiesis and thrombopoiesis during cancer development [19]. These factors notably include granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), thrombopoietin (TPO), interleukin-6 (IL-6), vascular endothelial growth factor (VEGF), basic fibroblast growth factor (b-FGF), and interleukin-1 (IL-1) [20]. Primary tumors secrete G-CSF and GM-CSF, which stimulate megakaryopoiesis in an endocrine manner and induce thrombopoiesis in people with cancer. Tumor-derived G-CSF also increases the number of neutrophils that can release neutrophil extracellular traps (NETs) and consequently promote thrombosis in people with lung cancer [21]. Many people with cancer also exhibit high serum levels of TPO and high platelet counts. TPO is a crucial cytokine that is secreted from the liver, kidneys, and bone marrow, and promotes megakaryocyte differentiation and proliferation, and resultant platelet generation after binding its receptor c-MPL, thus leading to activation of the Janus kinase (JAK)/signal transducers and activators of transcription (STAT) pathway [22]. In addition, cancer cells release multiple humoral factors and cytokines that upregulate hepatic TPO biosynthesis. The most prominent of these factors is the pleiotropic cytokine IL-6, a major mediator of inflammation and an activator of STAT3. And IL-6 overexpression promotes tumor development [23]. For instance, platelet microparticles (PMPs), released after platelet activation, promote the proliferation, survival, adhesion, and chemotaxis of hematopoietic cells through activating a vary of intracellular signaling cascades, such as the MAPK p42/44 and STAT pathways [24]. Activated platelets and excessive thrombin generation facilitate cancer-associated thrombocytosis in people with cancer.
2.2 Platelet activation and aggregation
The high risk of thrombosis in people with cancer arises from the ability of tumor cells to activate platelets and induce aggregation through direct and indirect mechanisms. Tumor-cell-induced platelet aggregation (TCIPA), which is associated with higher invasiveness and metastatic potential of tumors, has been demonstrated in a variety of tumors including pancreatic cancer [25, 26], colorectal cancer [27] and breast cancer [28]. Tumor cells release soluble platelet agonists from their membranes, including adenosine 5’-diphosphate (ADP), thromboxane A2 (TXA2), thrombin, and vWF, which directly activate platelets and stimulate thrombus formation [29]. Tissue factor (TF) is often expressed in tumor cells and tumor-derived microparticles; moreover, elevated serum levels of TF have been confirmed in multiple tumor types and cases of chemotherapy-induced thrombosis [30, 31]. Thrombin is a serine protease that converts fibrinogen to fibrin, and thrombin is the most potent platelet agonist acting on GPIb-IX-V and the protease-activated receptors (PAR) in platelets. Tumor cells also release ADP, which activates platelets via the purinergic G-protein-coupled P2Y1 and P2Y12 receptors. P2Y1 initiates ADP-induced platelet aggregation, and is responsible for platelet shape changes, whereas P2Y12 amplifies and stabilizes the aggregation response [10]. Tumor cells directly interact with platelets through receptors and ligands that mediate adhesion and aggregation, such as GPIb-IX-V, GPIIb/IIIa, and P-selectin [32]. Tumor cells also promote platelet activation through eliciting coagulation, which is triggered primarily by the release of TF from monocytes, thus providing an active surface for platelet adhesion and thrombus formation [33, 34].
2.3 Platelet education by tumors
Tumor-educated platelets are functional platelets in the blood circulation that express tumor-associated molecules [35]. The mechanisms through which tumors educate platelets include direct transfer of tumor-associated factors, tumor-induced changes in platelet RNA processing, and abnormal platelet production by MKs. Platelets may be continually stimulated and sequester molecules from tumor cells, thus resulting in changes in the platelet transcriptome, either via direct contact of membrane proteins with tumor cells or via extracellular molecules released by tumor cells. The platelets then express tumor-specific molecules, such as epidermal growth factor receptor (EGFR) vIII, echinoderm microtubule-associated protein-like 4 gene-ALK variant (EML4-ALK), Kirsten rat sarcoma viral oncogene homolog (KRAS), EGFR, phosphatidylinositol-4, 5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) variants, kallikrein-associated peptidase (KLK)2, KLK3, and neuropeptide Y (NPY) [36]. Beyond platelet mRNA content, platelets selectively take up plasma proteins, such as fibrinogen (Fg), VEGF, and serotonin (5-HT) into their granules, and release them after activation [37–39]. Furthermore, platelets have recently been shown to take up monocyte-derived cytokines, including IL-6, tumor necrosis factor (TNF-α), and IL-10; however, whether these cytokines can be stored and released remains to be determined [40]. Platelets also have a complicated endocytic machinery to absorb and store various proteins, including those derived from tumor cells. Tumor-cell-derived factors—including those that promote tumor growth and neovascularization, such as VEGF, PDGF, CXCL4/PF4, connective tissue-activating peptide III (CTAPIII), and thrombospondin-1 (TSP-1)—influence protein levels in platelets [41].
Modulation of platelet pre-RNA splicing is another important mechanism of platelet education by tumors. Despite their small size and lack of nuclei, platelets contain various types of RNA, including unspliced pre-RNA, mRNA, miRNA, ribosomal RNA, small nuclear RNA, transfer RNA, long noncoding RNA, antisense RNA, circular RNA, and functional spliceosomes for pre-RNA processing [42]. Various tumors have been shown to affect platelet mRNA content. Tumor cells transfer mRNA to platelets via released microvesicles and may alter the platelet transcriptome by regulating the splicing of pre-platelet mRNA [43]. Intriguingly, tumor-induced alterations in the platelet phenotype are considered important because of not only their possible functional consequences, but also their prognostic potential as biomarkers of tumor presence or progression [44, 45]. Tumors quantitatively alter platelet production and the number of mature platelets in the blood circulation; moreover, evidence indicates that tumors may modulate the platelet transcriptome and proteome by altering MKs [46]. Platelet content was previously believed to consist of “remnants” of progenitor MKs. However, MKs are now recognized to specifically sort RNA, proteins, and organelles which can be altered by various cancers into developing platelets.
2.4 Promotion of tumor invasion and metastasis by platelets
Tumor metastasis results in approximately 90% of tumor-associated mortality. Platelets participate in multiple steps of metastasis, including enhancing the invasive potential of tumor cells [47, 48]. Platelets have been reported to promote tumor invasion through various mediators and mechanisms. Platelet surface molecules (e.g., P-selectin, GPIbα, and αIIbβ3) and secreted factors from α-granules (e.g., TGF-β, lysophosphatidic acid (LPA), MMPs, and dense granules (e.g., serotonin, ADP, and histamine) all facilitate cancer invasion [49]. Tumor metastasis begins with tumor cell detachment from the primary tumor, a process requiring the tumor cells to undergo epithelial-mesenchymal transition (EMT), a process characterized by disrupted cell-cell interactions, expression of mesenchymal markers, and increased cell plasticity and stemness. Platelets upregulate mesenchymal markers (e.g., the Snail family transcriptional repressor-1, vimentin, N-cadherin, fibronectin, and MMP-2) and downregulate epithelial markers (e.g., E-cadherin and claudin-1), thus induing tumor cells to adopt an invasive, mesenchymal-like phenotype [50]. Activated platelets release TGF-β, which in turn causes tumor cells to adopt a pro-metastatic EMT phenotype [51]. Labelle et al. have demonstrated that platelet-derived TGF-β and direct contact between platelets and tumor cells synergistically activate the TGF-β/Smad and nuclear factor-κB (NF-κB) pathways in tumor cells, thereby enhancing EMT [52]. The platelet receptor C-type lectin-like receptor 2 (CLEC2) binds podoplanin expressed in tumor cells and subsequently initiates tumor cell cytoskeletal reorganization via ezrin/moesin-mediated intracellular signaling, thus enhancing the EMT phenotype and invasiveness of tumor cells [53]. Activated platelets also release autotaxin with lysophospholipase D activity from α-granules; these enzymes then catalyze the generation of LPA. Platelet-derived LPA stimulates IL-6 and IL-8 production in tumor cells, and eventually enhances osteolytic bone metastasis [54]. A recent study has indicated that the platelet-derived chemokine CCL5 and epidermal growth factor (EGF) stimulate tumor cell secretion of IL-8 by triggering Akt signaling [55]. Platelet-secreted CCL3 binds the CCR5 receptor on tumor cells and subsequently upregulates the membrane type I matrix metalloproteinase (MMP-1), possibly through activation of the NF-κB pathway, thereby promoting tumor cell invasion [56]. Platelets also participate in the formation of the pre-metastatic niche. Primary tumors secrete metastasis-associated proteins, which instruct target organs to recruit bone-marrow-derived cells, thus generating a pre-metastatic niche and promoting angiogenesis. Platelets regulate pre-metastatic communication [57]. For instance, platelet-derived chemokines (e.g., the CXC motif ligands CXCL5 and CXCL7) promote early metastatic niche formation by activating granulocyte-derived C-X-C chemokine receptor 2 (CXCR2).
Metastatic tumor cells must leave the primary tumor and intravasate into the blood flow; consequently, their survival before extravasation is critical for tumor metastasis. Although most circulating tumor cells (CTCs) are rapidly eliminated [58], blood components such as leukocytes and platelets can be mobilized to facilitate CTC survival and transit. Platelets are cells that CTCs encounter early in the blood circulation and are essential for CTC survival and transit in the blood circulation [59]. During the metastatic process, cancer cells encounter high shear stress and immune-system attacks on blood circulation. Cancer cells activate platelets by expressing tissue factors on their surfaces and stimulating various mediators, such as cathepsin G or thrombin. Activated platelets rapidly bind the surfaces of cancer cells and form a coating that protects tumor cells against shear forces and natural killer (NK) cells in the bloodstream. Furthermore, platelets decrease the recognition of cancer cells by NK cells through transferring “normal” major histone compatibility complex (MHC) class I antigens to tumor cell membranes, thus preventing NK cells from recognizing foreign cells, and alleviating NK-cell-mediated cytolytic activity and interferon (IFN)-γ production. Moreover, platelets secrete TGF-β, which decreases NK cell antitumor activity by downregulating NKG2D immunoreceptors. Tumor cells display “platelet receptors” such as αIIbβ3, αVβ3, or GP-Ibα, which facilitate direct tumor-endothelial and tumor-leucocyte interactions, and consequently promote tumor cell extravasation [48, 60]. For extravasation, CTCs must adhere to the luminal side of vascular endothelial cells and then break through the subepithelial ECM. The platelet membrane expresses multiple adhesive molecules including integrins (e.g., aIIbβ3, α6β1, and αvβ3), P-selectin, GPIb-IX-V, and immunoglobulin superfamily members (e.g., GPVI, FcγRIIa, and platelet/endothelial cell adhesion molecule-1 (PECAM-1)). These adhesion molecules on platelets interact with tumor cells and leukocytes, thereby facilitating tumor cell arrest within the vasculature [61, 62]. Meanwhile, platelets promote tumor cell adhesion to the vascular endothelium and binding to exposed subendothelial matrix proteins. Platelet integrin α6β1 directly interacts with tumor cell A Disintegrin and Metalloproteinase 9 (ADAM-9), which induces platelet activation and granule secretion, and promotes tumor cell dissemination [63]. In addition, platelet-derived microparticles deliver platelet-derived receptors such as CD41 to tumor cells, and facilitate tumor cell adhesion to the endothelium and fibrinogen [64]. GPVI on platelets is a specific collagen and fibrin receptor mediating platelet adhesion and tumor cell arrest in the vasculature through binding galectin-3 on tumor cells.
Molecules stored in platelet α- and δ-granules regulate vascular permeability. Activated platelets release VEGF, serotonin, platelet-activating factor (PAF), thrombin, ATP/ADP, hepatocyte growth factor (HGF), and fibrinogen, which increase vascular permeability and facilitate tumor cell transmigration [65, 66]. Tumor-cell-activated platelets release ATP from dense granules, and the ATP in turn interacts with the endothelial P2Y2 receptors, thus opening the transendothelial barrier and enabling tumor cells to pass through endothelial cell junctions into metastatic sites [67]. Interestingly, platelets store and release several exo-enzymes, such as MMPs, platelet hyaluronidase-2, and heparanase, which degrade collagen-rich ECM components and help tumor cells cross the subendothelial layer [65, 68].
2.5 Promotion of tumor growth and angiogenesis by platelets
Tumor growth requires angiogenesis, the formation of new blood vessels, to supply adequate nutrients, oxygen, and growth factors. Tumor angiogenesis arises from a complex network of interactions among tumor cells, the TME, and cells recruited from the bone marrow. Platelets supply a multitude of proangiogenic factors to tumors and promote the expression of proangiogenic factors by tumor cells, thereby regulating tumor angiogenesis and vascular integrity [69, 70]. Platelet α-granules contain numerous proangiogenic factors that directly or indirectly affect angiogenesis, such as VEGF, PDGF, basic fibroblast growth factor, and EGF. These factors not only promote angiogenesis and tumoral neovascularization, but also induce tumor growth. Interestingly, platelet α-granules also contain anti-angiogenic factors, such as angiopoietin-1 (ANGPT1), sphingosine 1-phosphate (S1P), thrombospondin-1 (TSP1), and endostatin. Platelets selectively release these factors, which facilitate or restrain angiogenesis in growing tumors, in a manner dependent on external stimuli. Stimulation of the receptor PAR1 contributes to the secretion of pro-angiogenic molecules; however, stimulation of the receptor PAR4 leads to the release of anti-angiogenic molecules. ADP-stimulated platelets release VEGF rather than endostatin, but thromboxane A2 (TXA2)-stimulated platelets release more endostatin than VEGF. Platelets can actively take up these factors and sequester them via endocytosis or megakaryocytes can selectively transfer a portion of proteins or mRNAs to platelets [71–73]. Platelets participate in early and advanced stages of angiogenesis, and help stabilize newly formed vessels. Platelets adhere to differentiated endothelial cells through their surface adhesion molecules, thus promoting endothelial cell proliferation and inducing angiogenesis in vivo. Platelet-released CXCL12/stromal-cell-derived factor (SDF-1) regulates revascularization by recruiting hematopoietic progenitors [34]. Platelets regulate not only tumor angiogenesis but also vascular integrity, thus preventing tumor hemorrhage via anti-angiogenic factors. Platelets maintain vascular integrity by secreting granules that contain ANGPT1 and serotonin, and platelets can stabilize tumor blood vessels by counteracting tumor cell-derived VEGF [74].
However, some studies have shown that platelet factor-4 (CXCL4/PF-4), which is released from α-granules of activated platelets, inhibits endothelial cell proliferation and migration, and thus displays anti-tumoral activity by inhibiting tumor growth and suppressing metastasis [75, 76].
3. ROLES OF PLATELETS IN THE TME
Various innate and adaptive immune cells, such as myeloid cells and lymphocytes, are major constituents of the TME that have been implicated in tumor progression and patient prognosis [77–79]. The composition and activity of immune cell populations in the TME dictate the state of the immune phenotype of the TME [78, 79]. Accumulating evidence indicates that platelets play major roles in the regulation of innate and adaptive immune responses [80]. Platelets, on their surfaces, express toll-like receptors (TLRs), which recognize pathogen- and danger-associated molecular patterns, similarly to leukocytes [80, 81]. After activation, platelets interact with immune cells and vascular cells directly and indirectly, through released inflammatory mediators such as the intracellular adhesion molecules (ICAM)-1, CCL2, CXCLs, IL-6, TNF-α, and TGF-β, thereby regulating the recruitment and activity of immune cells [80]. Regulation of the TME by platelets is discussed below.
3.1 Platelet regulation of leukocyte recruitment to the TME
Feng et al. have shown that depletion of platelets with antibodies to GPIbα significantly decreases the infiltration of bone-marrow-derived cells in B16-F10 tumor isografts, whereas platelet infusion has an opposite effect, and deficiency in α-granule secretion completely abolishes platelets’ effects [82]. In a diethylnitrosamine-induced mouse model of hepatocellular carcinoma, blocking platelet activation by clopidogrel significantly decreases the density of macrophages in tumors [83]. Platelet-released chemokines, such as CCL2, IL-8, CXCL5, CXCL7, and CXCL12, are powerful recruiters of leucocytes [84, 85]. Macrophages have CXCR4 positivity and are recruited to metastatic tumors by platelet-derived CXCL12 [85]. Gil-Bernabé et al. [86] have shown that platelet clotting induced by tumor-cell-expressed TF enhances macrophage recruitment in mouse B16-F10 lung metastatic tumors. In the same lung metastasis model, Labelle et al. have found that platelet depletion completely inhibits granulocytic cell recruitment to early metastatic sites, and the effects of platelets are attenuated by blocking platelet-secreted CXCL5 and CXCL7 [57]. In addition, Läubli et al. [87] have shown that platelets, together with polymorphonuclear leukocytes/monocytes, increase monocyte infiltration in lung metastatic colorectal cancer tumors in mice via inducing CCL5 secretion from endothelial cells, thus revealing an indirect route through which platelets promote leukocyte recruitment to the TME. Platelets have also been reported to promote monocyte recruitment in the TME through induction of CCL2 expression in tumor cells [85].
3.2 Inhibitory regulation of the TME by platelets
Macrophages in the TME, i.e., tumor-associated macrophages (TAMs) primarily acquire an immunosuppressive, M2-like phenotype. High presence of this phenotype in both the tumor tissues and the blood has been associated with poor patient prognosis [88–90]. Platelets are the major source of TGF-β in the TME; this cytokine is a potent inducer of M2-like TAMs [91, 92]. In mouse models of hepatocellular carcinoma, clopidogrel (a P2Y12 receptor inhibitor) significantly increases the expression of M1-like anti-tumor macrophage markers, such as IL-1, TNFα, and inducible nitric oxide synthase (iNOS) [83]. Analogously to TAMs, tumor-associated neutrophils (TANs) adopt primarily a tumor-promoting (N2-like) phenotype in the TME, in a process largely driven by TGF-β [90, 93]. NK cells and cytotoxic T lymphocytes (CTLs) are effector cells that destroy tumor cells through multiple mechanisms, including the secretion of effector cytokines, such as TNF-α and IFN-γ, and the release of cytotoxic granules [94, 95]. Protection of CTCs against NK-cell-mediated immunosurveillance by platelets has been observed in mouse models of lung metastasis involving multiple tumor cell lines including melanoma and fibrosarcoma [96]. NK cell activation is triggered by target cells with low or absent expression of MHC class I (missing-self) and/or overexpression of ligands for activating NK receptors, such as natural killer group 2 member D (NKG2D), receptor activator of NF-κB (RANK), and glucocorticoid-induced tumor necrosis factor receptor (GITR) (induced self) [94]. Platelet co-incubation with tumor cells leads to the transfer of platelet-derived MHC class I molecules, RANK ligands, and GITR ligands to tumor cells, thus impairing NK cell antitumor reactivity and IFN-γ release [60, 97, 98]. Activated platelets induced by tumor cells have also been found to decrease the levels of the NKG2D ligands MHC class I polypeptide-associated sequence A (MICA) and MICB on tumor cell surfaces through the sheddases A Disintegrin and Metalloproteinase 10/17, thereby inhibiting NKG2D-mediated NK cell recognition of tumor cells [99]. In addition, platelet-secreted TGF-β impairs IFN-γ production by, and degranulation of, NK cells through down-regulation of NKG2D in NK cells [100]. Similarly, dabigatran etexilate (a thrombin inhibitor) significantly increases the infiltration of NK cells and CD8+ T cells in mouse MC-38 tumors by systemically decreasing active TGF-β1 [101]. Upregulation of immune checkpoint proteins such as programmed death ligand 1 (PD-L1) and CTL-associated antigen 4 (CTLA-4) is a well-documented mechanism through which tumors suppress CTL-mediated immune responses [102]. Hinterleitner et al. [103] have shown that co-incubation of platelets with PD-L1+ NCI-H226 and NCI-H460 tumor cells increases PD-L1 expression on platelet surfaces. PD-L1+ platelets obtained from patients with non-small cell lung cancer have been observed to suppress CD8+ T cell activity, thus decreasing the release of IFN-γ and TNF-α. Moreover, TGF-β released from platelets converts CD4+ T cells into regulatory T cells (Tregs), which destroy activated T cells in a manner dependent on granzyme B [91].In a mouse model of colitis-associated cancer, clopidogrel treatment has been found to inhibit the infiltration of MDSCs into tumors [104]. Platelets isolated from mice with colitis-associated cancer have also been shown to enhance splenic MDSC-mediated inhibition of T-cell proliferation. These findings collectively suggest that platelets promote the establishment of the suppressive TME through direct or indirect interactions with immune cells.
3.3 Stimulatory regulation of the TME by platelets
Platelet interactions with immune cells in the TME are complex and appear to have both suppressive and stimulatory effects. Local treatment of platelet-rich fibrin patch (PRF-P) inhibits the recruitment of Tregs to the glioma microenvironment by the secretion of soluble CD40 ligand (sCD40L) [105]. Although platelets are not the only source of sCD40L in PRF-P, this finding supports the possible involvement of platelets in antitumor immunity activation. Plantureux et al. [106] have shown that the interaction of platelets with the colorectal cancer cell lines HT-29 and CT-26 in the absence of plasma induces the production of CD41+/Epcam+ (surface markers of platelets and tumor cells) microparticles in a manner dependent on cadherin 6. The CD41+/Epcam+ microparticles contain RANTES, macrophage migration inhibitory factor (MIF), CXCL-12, and IFN-γ, thus enhancing the recruitment of M1-like macrophages in the CT-26 cell TME, and resulting in increased expression of iNOS and Arg1. The interaction of macrophages plus platelets with tumor cells has been confirmed to result in cell-cycle arrest in vitro and decreased tumor growth in vivo. This finding appears to contradict those from a study by Pavlović et al. [83], in which the suppression of platelet activity by clopidogrel increased the expression of M1 macrophage markers, such as IL-1, TNF-α, and iNOS, in the tumors of mice with hepatocellular carcinoma. Clopidogrel is an inhibitor of the P2Y12 receptor, which targets primarily ADP-mediated TCIPA. In contrast, Plantureux et al. [106] have observed that the production of CD41+/Epcam+ microparticles is induced by the interaction of platelets with tumor cells through cadherin-6, independently of TCIPA. The different mechanisms of interaction between platelets and tumor cells may be a possible reason for these contradicting findings, and other differences among the tumor types used may also be nonnegligible factors.
4. PLATELET-LEUKOCYTE INTERACTIONS IN INFLAMMATION AND AUTOIMMUNE DISEASES
As described above, the immunosuppressive TME might exhibit immune stimulation and inflammatory responses in response to the immunostimulatory effects of multiple anti-tumor therapies, which convert immune-excluding cold tumors to immune-infiltrated hot tumors. Platelets are key components of the tumor immune microenvironment that play important roles in tumor immunosuppression mechanisms by directing and/or assisting immune cells. Thus, the roles of platelets in remodeling of the suppressive tumor immune microenvironment must be considered, despite limited evidence emphasizing the immune effects of platelet-leukocyte interactions in tumor damage. Nonetheless, the involvement of platelet-leukocyte interactions in numerous pathological conditions, including acute coronary syndrome, sepsis, ulcerative colitis, asthma, and rheumatoid arthritis, has been well described. Increased platelet-leucocyte aggregates within the circulation and/or locally at the sites of inflammation have been observed in the above inflammatory/autoimmune diseases and found to be associated with disease progression, thus serving as potential biomarkers of disease severity [123–125] In recent years, as the immune regulatory role of platelets has increasingly been recognized, studies have focused on the consequences of platelet-leukocyte interactions, including the recruitment and activation of leukocytes, which enable the execution of leukocytes’ pro-/anti-inflammatory functions.
4.1 Platelet-leukocyte interactions promoting inflammation and tissue damage
During tissue inflammatory damage, activated vascular endothelial cells and platelets produce enormous amounts of adhesion molecules, such as P- and E-selectin, that allow cells to stick to one another and function as a bridge that tethers and anchors leukocytes, thus initiating leukocyte recruitment to inflammation sites [107, 126] The importance of platelet P-selectin has been verified in multiple disease models by transferring P-selectin-deficient platelets into mice, in which impaired leukocyte recruitment has been observed to be accompanied by diminished tissue inflammatory damage [120, 127–131]. In addition to P-selectin-mediated direct contact, platelets upregulate endothelial selectin expression by secreting 5-HT, thereby increasing neutrophil adhesion to LPS-stimulated endothelium [132]. Using multi-channel intravital microscopy to observe inflamed microvessels of the cremaster muscle in mice, Zuchtriegel et al. [129] have shown that intravascularly adherent platelets not only capture neutrophils and inflammatory monocytes via CD40-CD40L-dependent interaction, but also promote firm adhesion and intravascular crawling of neutrophils and inflammatory monocytes in a P-selectin dependent manner, thus ultimately guiding them to the exit points of transmigration. A lack of platelet CD40 leads to decreased plaque area, and infiltration of macrophages and neutrophils, in atherosclerotic mice injected with apolipoprotein E deficient platelets [133]. In mouse models of acute lung injury and permanent middle cerebral artery occlusion, neutrophils recruited to damaged vessels can also use PSGL-1 clusters to scan for the presence of activated platelets; subsequently, neutrophils begin to organize other receptors (e.g., CXCR2 and Mac-1) required for intravascular crawling and transmigration [134]. Indeed, platelet-derived CXCL4 has been demonstrated to enhance the infiltration of neutrophils and inflamed tissue damage by upregulating the plasma and tissue levels of CXCL1 and CXCL2 in mice with sepsis and acute pancreatitis; these effects are neutralized by inhibition of CXCR2 [135, 136]. Schuhmann et al. [137] have also shown that inhibition of platelet GPIb limits the local inflammatory response in the ischemic brain in mice with transient middle cerebral artery occlusion; consequently the numbers of infiltrating macrophages and T cells decrease, possibly because GPIb contains a binding site for Mac-1. Accordingly, in mouse models of house-dust-mite-induced asthma or major Leishmania infection, platelet-derived Dkk1 has been found to facilitate leukocyte infiltration, including that by CD4+ T cells, neutrophils, and eosinophils, to sites of inflammatory stimulation [138]. Platelet-derived sCD84 accumulates in the ischemic cerebral microvasculature of mice with transient middle cerebral artery occlusion and exerts proinflammatory effects by acting on CD4+ T cells, thus promoting their recruitment and aggravating cerebral ischemia/reperfusion injury [139]. Notably, CD40L-bearing T cells have been reported to induce platelet activation and platelet-CCL5 release in a manner dependent on a CD40-CD40L costimulatory mechanism [140]. This finding is supported by an in vivo study in which CD4+ T cells have been found to enhance the interactions between platelets and endothelial cells, and the migration of neutrophils in the postischemic hepatic microcirculation through similar costimulatory mechanisms [141]. Activated platelets and released platelet-CCL5 might in turn enhance CD4+ T cell recruitment, thereby creating a positive feedback loop that further amplifies leukocyte recruitment to sites of immune reactivity [140].
Beyond promoting various steps in leukocyte recruitment, platelets modulate the immunological functions of leukocytes. P-selectin-PSGL-1 and GPIb-Mac-1 are also key players with pleiotropic actions in the regulation of leukocyte immune functions. Under a subthreshold dose for M1 macrophages polarization of LPS stimulation, platelets promote macrophage phenotypic and functional polarization toward a proinflammatory profile, in a manner dependent on GPIb-Mac-1 [40]. Thus, platelets protect mice against septic shock early during sepsis development by inducing iNOS-positive M1-like macrophages, which enhance bacterial clearance. ROS produced and release by leukocytes destroy invading pathogens and trigger tissue damage, owing to their extreme cytotoxicity [126, 142]. P-selectin-PSGL1 binding mediates a direct interaction between platelets and neutrophils, thus increasing the efficiency of neutrophil ROS generation [107]. Moreover, activated platelets have been reported to promote ROS production by monocytes via dissociating C-reactive protein in its proinflammatory monomeric form [108]. In a mouse model of myocardial reperfusion injury, depletion of platelet 5-HT significantly decreases ROS release and secretory granules containing MPO in neutrophils. Similarly to ROS, MPO enhances inflammation and tissue damage [109]. The compound 5-HT has been reported to prolong monocytes’ lifespan by preventing their spontaneous apoptosis and enhancing their ability to produce proinflammatory cytokines, including TNF-α and IL-6, after stimulation with LPS; therefore, platelet 5-HT may amplify allergic inflammation [110]. In addition, platelets have been found to enhance phagocytosis of periodontitis-associated bacteria by neutrophils in a TLR2-dependent manner [143]. Moreover, TLR4 activation on platelets during severe sepsis induces NET formation and liberation [111]. NETs have a netlike configuration decorated with DNA, antimicrobial molecules such as histones, defensins, and various neutrophil proteases, which are released by neutrophils when phagocytosis is frustrated, which enhances the ability to capture and kill pathogens [112, 126, 142]. Because platelet-neutrophil-mediated NET formation requires excessive activation of neutrophils, substantial endothelial and tissue damage may result [111, 112]. Moreover, NETs activate platelet through histones; hence, platelet-neutrophil-mediated NETs might promote inflammation and tissue damage through positive feedback loops [112, 113]. Petersen-Uribe et al. [114] have shown that platelet-derived PCSK9 promotes monocyte differentiation into macrophages/foam cells during atherothrombosis. In patient atherosclerotic tissues, the authors have observed high macrophage CD68-immunoreactivity in PCSK9-and platelet-positive areas, thus suggesting a possible role of platelets in promoting atherosclerosis-favoring inflammation. In the setting of asthma and allergic diseases, such as atopic dermatitis and allergic rhinitis, TSLP from the epithelium might activate the transformation of epithelial and dermal DCs to TSLP-DCs. Activated platelets promote the maturation of TSLP-DCs and the production of the Th2-attracting chemokine CCL17 in vitro through RANK-RANKL and CD40-CD40L pathways, in which Th2 cells are crucial for the initiation and maintenance of allergic inflammation [118]. In addition to regulating the interactions between APCs and lymphocytes, platelets have been reported to influence lymphocyte differentiation, activation, and cytokine production via various chemokines or direct cell contact, in which the CD40-CD40L pathway is a key mechanism [120, 121]. In mouse experimental asthma models, platelet CD40L has also been observed to directly inhibit the generation of Tregs both in vitro and in vivo, thus providing a mechanistic explanation for platelets’ promotion of Th2-type inflammation in asthma [122].
The above findings regarding the multitude of interactions between platelets with leukocytes compellingly suggest that platelets crucially enhance the innate and adaptive immune responses in the context of inflammation and autoimmune diseases ( Table 1 ).
Main mediators of interactions between platelets and tumor cells or immune cells.
| Platelet-derived mediators | Interactions | Functions | Reference |
|---|---|---|---|
| α6β1 | ADAM9 on tumor cells | [61] | |
| GPIbα | vWF on tumor cells | [33] | |
| TLR-4 | HMGB1 on tumor cells | [61] | |
| P-selectin | PSGL-1 on tumor cells | [61] | |
| CLEC-2 | Podoplanin on tumor cells | [50] | |
| GPVI | Galectin-3 on tumor cells | [64] | |
| GPIIb/IIIa | Cadherin 6 on tumor cells | [34] | |
| P-selectin | PSGL-1 on neutrophils, monocytes, and T cells | [107, 126, 129, 144] | |
| GPIb | Mac-1 on neutrophils, macrophages, and T cells | [40, 107, 135–137] | |
| CD40 | CD40L on neutrophils, monocytes, T cells, and DCs | [118, 120, 121, 133, 140] | |
| 5-HT | 5-HT 1/4/6/7 receptor subsets on neutrophils and monocytes | [109, 110, 132] | |
| CXCL4 | CXCR2/3 on neutrophils Unknown on T cells | [135, 136, 144] | |
| CCL5 | CCR5 on T cells | [140] | |
| Soluble CD84 | CD84 on T cells | [139] | |
| TLR2 | Unknown on neutrophils | [143] | |
| TLR4 | Unknown on neutrophils | [111] | |
| TGF-β | Unknown on T cells | [145] |
4.2 Platelet-leukocyte interactions orchestrating the resolution of inflammation
The functions of platelet-leukocyte interactions appear to be multifaceted and intimately involved in negative feedback regulation of a finely tuned immune system, to guard against excessive inflammatory responses. During the early stages of mouse experimental autoimmune encephalitis, signs of platelet activation/degranulation have been observed, in which soluble factors such as 5-HT and CXCL4 exhibit proinflammatory properties by stimulating the proliferation and differentiation of pathogenic Th1, Th17, and IFN-γ+/IL-17+ CD4+ T cells. However, in advanced stages of multiple sclerosis and experimental autoimmune encephalitis, activated platelets display enhanced ability to form aggregates with CD4+ T cells, resulting in decreased T cell activation [144]. Depletion of platelets leads to impairments in protective CD4+ Treg activation early after trauma in a mouse model of burn injury [146]. Similar studies in sepsis mouse models have shown that blocking the P2Y12 receptor with clopidogrel alleviates TGF-β elevation in the plasma and restrains Treg population growth in the spleen during sepsis [145]. Using an immune tolerance mouse model, Hotta et al. [147] have shown that platelet depletion reverses the increase in Tregs in the inflamed skin and draining lymph nodes of mice via TGF-β release, and significantly enhances the contact hypersensitivity response tolerance of cutaneous inflammation. Furthermore, in a recent study, platelets have been observed to recruit neutrophils into the lungs during the onset of bacterial-induced pneumonia, and to recruit Tregs and transcriptionally reprogrammed alveolar macrophages to an anti-inflammatory phenotype during the resolution phase. The mechanisms of this observation have been postulated to involve cleavage of PSGL-1 on neutrophils by the sheddase ADAM8, thus enabling preferential binding of platelets to Treg cells during the resolution phase [148].
In summary, platelets have been found to have active roles in the TME that are crucial in tumor-induced or tumor-associated immunosuppression ( Figure 1 ). Platelets also initiate and accelerate inflammatory processes and immune responses in the context of inflammation and autoimmune diseases. Increasing knowledge and deeper understanding of theses aspects are expected to boost the development of anti-tumor therapeutic strategies that subvert platelets’ immunosuppressive properties and/or exploit their immunostimulatory properties to achieve better efficacy.
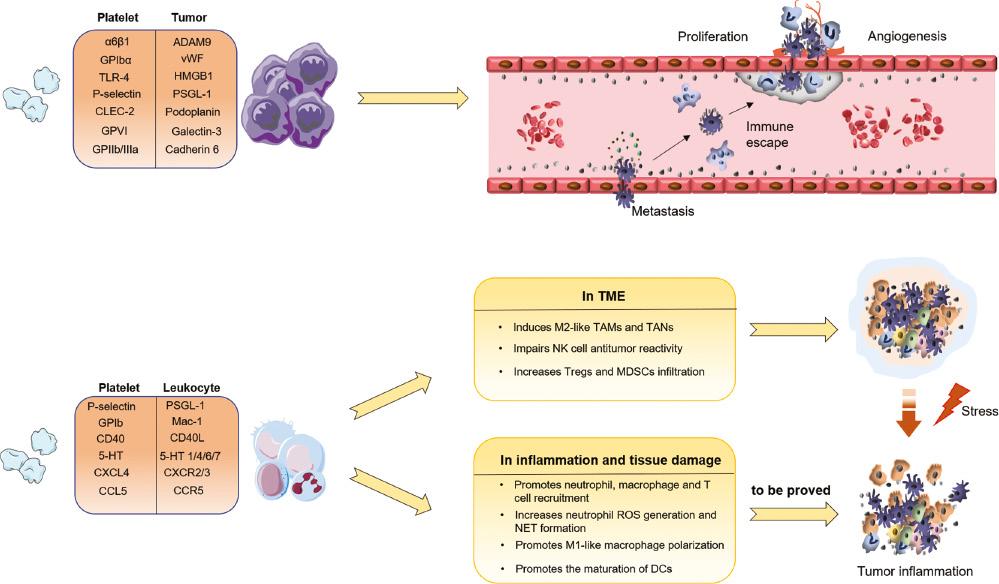
Model of the possible involvement of platelet-tumor cell and platelet-immune cell interactions in tumor development and the TME.
Platelet-tumor cell interactions contribute to tumor proliferation, angiogenesis, and metastasis. Platelet-leukocyte interactions also play crucial roles in the mechanisms of tumor-associated immunosuppression. Platelet-leukocyte interactions can initiate and accelerate inflammatory processes and immune responses in the context of inflammation and autoimmune diseases.
5 PLATELETS FOR ANTITUMOR DRUG DELIVERY
5.1 Unmodified platelets as carriers for drug delivery
The goal of drug delivery is to enhance both therapeutic safety and efficacy, and the use of cells as delivery vehicles is a promising approach. Most vehicle cells used to date are immune cells, e.g., monocytes, macrophages, dendritic cells, neutrophils, and platelets, which have a natural propensity to home to sites of tissue injury and inflammation, such as cancer, and can cross tissue barriers such as the blood brain barrier. Among the candidate carrier cells for drug delivery, platelets have several unique advantages. For example, platelets take up and stably store large amounts of both endogenous and exogenous molecules including antitumor agents [149]. Healthy platelets usually maintain circulation in the bloodstream for 8–10 days, thus resulting in long half-lives of drug cargo. Platelets release the contents stored in their granules after activation by tumors, thereby increasing the tumor-targeting capabilities of platelet-mediated delivery. Sarkar et al. have shown that platelets take up doxorubicin (DOX) and release the agent after activation, and DOX-loaded platelets have been found to induce tumor cell toxicity with higher efficiency than free DOX in both in vitro and in vivo models [150]. Xu et al. have loaded DOX in platelets to treat mouse lymphoma models [151]. The DOX-loaded platelets have been found to maintain integrity and biological functions; to have high DOX loading capacity and encapsulation efficiency; to release DOX at tumor sites in a pH-triggered manner; and to increase DOX uptake by Raji tumor cells, thus enhancing growth inhibition and apoptosis of Raji tumors, and alleviating normal tissue damage. In our laboratory, we have found that platelets have a low loading capacity for small-molecule drugs, e.g., DOX and chlorin e6 (Ce6), and platelets loaded with these drugs exhibit substantial spontaneous cargo discharge, thus limiting the utility of platelets as drug carriers. This problem has been overcome through using nanoparticles (NPs) as the primary drug vehicles and using platelets as secondary carriers of the drug-loaded NPs. Our laboratory has recently demonstrated platelet-mediated tumor-targeted drug delivery using DOX attached to nanodiamonds as a model drug [149]. The platelet drug carriers have the following features: 1) high loading capacity and efficiency, 2) good tolerance of the payload drug, 3) stable cargo retention and no cargo release without stimulation, 4) long blood circulation times, and 5) good tumor distribution and tumor-activated cargo unloading. These features enable a much higher therapeutic potency than that of DOX, with little systemic toxicity. An artificial stimulus, e.g., ROS generated by photodynamic/sonodynamic therapy or heat generated by photothermal therapy also causes platelet activation and cargo release, thus providing an advantage of highly controllable drug release in platelet-mediated delivery. On this basis, we have invented a drug delivery strategy of using platelets with photo-controlled release properties [152]. To provide a proof of concept for this strategy, we have prepared the delivery device BNPD-Ce6@Plt by loading mouse platelets with a nano-photosensitizer (BNPD-Ce6) consisting of Ce6 loaded in boron nitride NPs with a surface coating of polyglycerol and DOX. Irradiation with an 808 nm laser induces ROS generation in the BNPD-Ce6@Plt, which have been found to exhibit rapid activation, aggregation, and discharge of BNPD-Ce6 into co-cultured GL261 glioma cells, which in turn display pronounced photodynamic toxicity and cell death. In vivo, laser irradiation of intracranial GL261 tumors after i.v. injection of BNPD-Ce6@Plt has been observed to lead to extensive distribution and accumulation of BNPD-Ce6 in tumors, which display massive tissue necrosis after additional laser irradiation. A photodynamic therapy regimen of intravenous BNPD-Ce6@Plt injections followed by tumor laser irradiation has been found to markedly suppress the growth of intracranial GL261 tumors and significantly prolong host survival. Zhang et al. have also provided strong support for the use of platelets as drug carriers with photo-controlled release properties [153]. The authors have constructed a drug delivery device (IRDNP-PLT) based on platelets with encapsulated DOX NPs and IR-820, a fluorescent photothermal agent. The IRDNP-PLT device enables controlled release of DOX NPs by IR-820-mediated photothermal activation. Near-infrared irradiation of subcutaneous 4T1 tumors in mice after i.v. injection of IRDNP-PLT has revealed highly targeted and enhanced antitumor drug delivery, and synergetic photo-chemotherapeutic efficacy. These findings, together with ours, compellingly suggest that intact platelets can serve as drug carriers with active tumor-targeting properties, which can be greatly enhanced through external manipulation.
Like naïve platelets, platelets harboring encapsulated drugs are cleared by the reticuloendothelial system, which comprises tissue macrophages and blood-borne monocytes. Recruitment and extravasation of circulating neutrophils are largely platelet dependent; thus, platelets might also be exploited to deliver agents that regulate or modulate the functions of monocytes, macrophages, and neutrophils, which are essential to anti-tumor immunity. Platelets are particularly amenable to carrying agents whose actions target the nucleus, because platelets are anucleated and thus remain unaffected when carrying these agents [154].
5.2 Platelet engineering for drug delivery
Tropism toward inflammation, adhesion properties, and dynamic changes after activation make platelets multifunctional platforms suitable for drug delivery and immune regulation. Platelets can also be directly modified through surface engineering, owing to the abundance of various binding sites on their plasma membranes [155]. Platelets can be harvested in sufficient quantities from patients with relative ease and then infused back into the same patients after in vitro modification. This ability is in line with the trends of precision medicine and personalized therapy, and eliminates immunogenicity-associated adverse effects. Engineering platelets for tumor immunotherapy appears to have great promise, by increasing immune cell penetration and activation in tumors, thereby stimulating robust antitumor immunity. Wang et al. have used a facile method to conjugate antibodies to PD-L1 (aPD-L1) to the surfaces of mouse platelets via a maleimide linker, to decrease tumor local recurrence and metastasis [156]. The aPD-L1-conjugated platelets (P-aPD-L1) are recruited to surgical wound sites because of their natural tendency to home to sites of tissue damage and inflammation. Consequent activation of P-aPD-L1 at wound sites leads to the release of many PMPs with aPD-L1 presented on the surface. Consequently, aPD-L1 accumulate at tumor resection sites, and T cell infiltration and activation occur at the remaining tumor resection site, thus eventually preventing tumor recurrence after surgery. P-aPD-L1 also interacts with CTCs and inhibits tumor metastasis. Han et al. have used FDA-approved poly (lactic-co-glycolic) acid-coated indocyanine green (PLGA-ICG) as a model photothermal agent in tandem with P-aPD-L1 to treat tumor recurrence [157]. They have demonstrated that thermal ablation creates inflammatory sites in tumor tissue that exhibit enhanced recruitment of aPD-L1-conjugated platelets. Activated aPD-L1-conjugated platelets then release the aPD-L1, which block PD-L1 on both tumor cells and antigen-presenting cells, thereby promoting T-cell infiltration and activation, and inhibiting residual tumor growth and metastasis. Beyond promoting immune checkpoint inhibitor therapy, engineered platelets can be used to combine immunotherapy with other therapies to achieve synergistic antitumor efficacy. Engineered platelets delivering therapeutic agents to tumors work with CAR-T cells, thereby overcoming the immunosuppressive TME, T cell exhaustion, and the occurrence of graft-versus-host disease [158]. After activation, platelets serve as bioresponsive cells that release aPD-L1 intratumorally, thereby sustaining CAR-T-cell-mediated antitumor activity and preventing T cell exhaustion [159].
However, it is costly to conjugate PD1 or aPD-L1 to the surface of platelets, and relatively large amounts of antibodies are required to obtain sufficient amounts of therapeutic materials. In addition, platelets that stably express PD-1 are fairly difficult to obtain; therefore, more advanced or refined techniques must be developed to generate engineered platelets in sufficient quantities. To overcome the technical challenges in genetic engineering of anucleated platelets, substantial effort has been devoted to using precursor cells, such as MKs, to produce platelets with the desired functions. Zhang et al. have recently genetically engineered MKs progenitor cells to produce platelets expressing PD-1 [160]. They have demonstrated that the PD-10expressing platelets and derived microparticles accumulate in the TME and block the PD-L1, thus inhibiting the activity of the immunosuppressive Tregs and enhancing the activity of CD8+ T lymphocytes at tumor resection sites. Furthermore, they have loaded low-dose cyclophosphamide (CP) into PD-1-expressing platelets, which have been found to deplete Tregs and increased reinvigorated CD8+ T lymphocyte cells in the post-surgery TME. Although viability and desired biological functions can be well preserved in genetically or chemically engineered platelets, undesirable effects on other platelet functions and the long-term behavior of the engineered platelets require thorough evaluation.
5.3 Platelet membrane camouflage for drug delivery
Numerous NP-based delivery systems have been devised for tumor chemotherapy. NP encapsulation can enable controlled drug release, improved antitumor drug distribution/accumulation, increased tumor tissue permeability, and diminished systemic toxicity and adverse effects [161, 162] Despite substantial progress, some problems remain to be addressed, such as nanotoxicity, unsatisfactory targeting capability, unfavorable immune responses, and short circulating times in the bloodstream. To address these issues, various cell membranes have been introduced onto NP surfaces to form biomimetic cell membrane camouflaged drug delivery devices. These devices have cell surface proteins whose biological functions are retained, without the disadvantages associated with using live cells, such as activation, cell toxicity, and low drug loading capacity. Among the many candidate cell membranes, the platelet membrane is particularly attractive [163, 164]. Platelet membranes with various integrated nanomaterials display distinct properties of platelets, e.g., long blood circulation times, cell adhesion, interaction with immune cells, and targeting of tumor cells, as well as additional features such as biocompatibility, biodegradability, and translocation across biological barriers. Coating NPs with platelet membranes also avoids undesired bio-distribution, unexpected activation-associated drug toxicity, and diminished efficacy [151, 152]. The combination of platelet membranes with chemotherapy, or photothermal or photodynamic therapy, may have synergistic antitumor effects. Pei et al. have developed platelet-membrane-coated poly-lactic-co-glycolic acid NPs (PM-NPs) loaded with IR780, a photothermal agent with tumor-targeting and superior optical properties, and the chemotherapeutic agent DOX [165]. The PM-NPs have good stability, prolonged blood circulation time, near-infrared-enhanced tumor accumulation, and active targeting of cancer cells, thus substantially enhancing anti-tumor photothermal therapy efficiency. Platelet membrane camouflaged devices have also been used to target CTCs to suppress tumor metastasis. CTCs induce platelet activation, fibrin deposition, and local thrombosis, thus protecting against immune attack. Therefore, Li et al. have coated silica (Si) particles with membrane-derived vesicles (PMDVs) from activated platelets and conjugated tumor necrosis factor-associated apoptosis-inducing ligand (TRAIL) to platelet-membrane-covered Si particles [166]. The resultant PMDV-Si particles exhibit diminished phagocytic clearance, owing to the presence of membrane proteins and glycans from platelet membranes, such as CD47. The PMDV-coated Si particles adhere to fibrin under flow, and the TRAIL-conjugated PMDV-coated Si particles induce tumor cell apoptosis via the tumoricidal activity of immune cells. Eventually, the TRAIL-conjugated PMDV-Si particles kill CTCs in the lung vasculature and decrease lung metastasis. The combination of platelet membrane camouflaged drug delivery with immunotherapy has also been shown to enhance antitumor immunity in solid tumors. Resiquimod (R848), a TLR agonist, has been coated with platelet membranes (PNP-R848) to achieve tumor-localized delivery and elicit antitumor immune responses [167]. The biomimetic platelet-derived membrane of PNP-R848 displays enhanced affinity to tumor cells and improved tumor retention. PNP-R848 promotes the activation of APCs in draining lymph nodes, thus enhancing T cell infiltration in tumors, and ultimately leading to tumor eradication, metastasis suppression, and generation of memory T cells that thwart subsequent tumor re-challenge. These findings suggest the potential of platelet membrane camouflaged delivery for tumor immunotherapy. Although the multifaceted biointerfacing supported by platelet membrane cloaking technology provides a new approach for developing functional NPs for targeted tumor therapy, biomembrane-derived biomimetic nanomedicines must address the issue of reproducibility for membrane protein quantities and their large-scale production. Moreover, the integrity of the platelet membranes, and the integrity and functions of the NP core in the blood are not easily characterized. Although platelet membrane-disguised NPs are generally accepted to be safe, little information is currently available regarding their long-term biocompatibility and toxicity profiles.
5.4 Platelet-derived microparticles for antitumor drug delivery
Platelets activated by tumor cells generate PMPs that are released into the blood. They are formed by cell membrane budding and range from 0.1 to 1 μm in size. Platelet-derived microparticles, also known as microvesicles, participate in cell-to-cell communication. Microparticles deliver payloads of lipids, proteins, miRNAs, mRNAs, and non-coding RNAs into tumor cells, thereby modulating gene expression and functions [168]. Recent studies have shown promising results in using microparticles as novel therapeutic vehicles for drug delivery. Microparticles are poorly immunogenic, shield therapeutic cargoes from rapid degradation in vivo, and can cross biological barriers such as the blood-brain barrier. Kailashiya et al. have engineered human platelets to generate microparticles loaded with DOX and other agents through a top-down approach; the particles have successfully targeted leukemia cells and have good biocompatibility [169]. Moreover, PMPs can carry multiple drug payloads, have a long shelf life, and can be harvested in large quantities in short periods of time. Importantly, PMPs exhibit remarkably higher toxicity toward cancer cells than free drugs, and show less escape into extravascular spaces. PMP-mediated delivery, compared with administration of free drugs, has been found to achieve significantly higher drug content in cancer cells of patients with leukemia.
Despite the promise of nanovesicle drug delivery, several challenges must be addressed before this delivery method can be used clinically. All cell types isolated from the blood must undergo a rigorous sterilization process to avoid the risk of infection. Moreover, if blood of non-autologous origin is used, blood screening is required after blood-type matching, to ensure maximum compatibility.
6. CONCLUSIONS AND OUTLOOK
In summary, platelets have emerged as promising and versatile candidates for the delivery of tumor-targeted therapies, owing to their unique, advantageous biological properties including stable and high drug-loading capacity, amenability to modification and engineering, long blood circulation time, tumor homing, ability to cross biological barriers, and tumor- or stimulus-induced activation and aggregation ( Table 2 ). Furthermore, platelets have close interactions with tumor cells and tumor-associated immune cells, both in the blood circulation and at the local tissue level, and thereby have a fundamental influence on the immune system in the tumor context. Hence, platelets have emerged as both intriguing therapeutic targets and powerful, maneuverable tools for the treatment of tumors ( Figure 2 ).
Advantages and disadvantages of platelet-based anti-tumor drug delivery systems.
| Strategies | Reported tumor types | Advantages | Disadvantages | Reference |
|---|---|---|---|---|
| Unmodified platelets as carriers for drug delivery | Lymphoma, glioblastoma, and 4T1 tumors | [151–153] | ||
| Platelet engineering for drug delivery | Melanomas and triple-negative breast carcinomas |
| [156–158] | |
| Platelet membrane camouflage for drug delivery | Breast cancer and colorectal adenocarcinoma |
| [165–167] | |
| Platelet-derived microparticles for antitumor drug delivery | Leukemia |
| Risk of contamination | [169] |
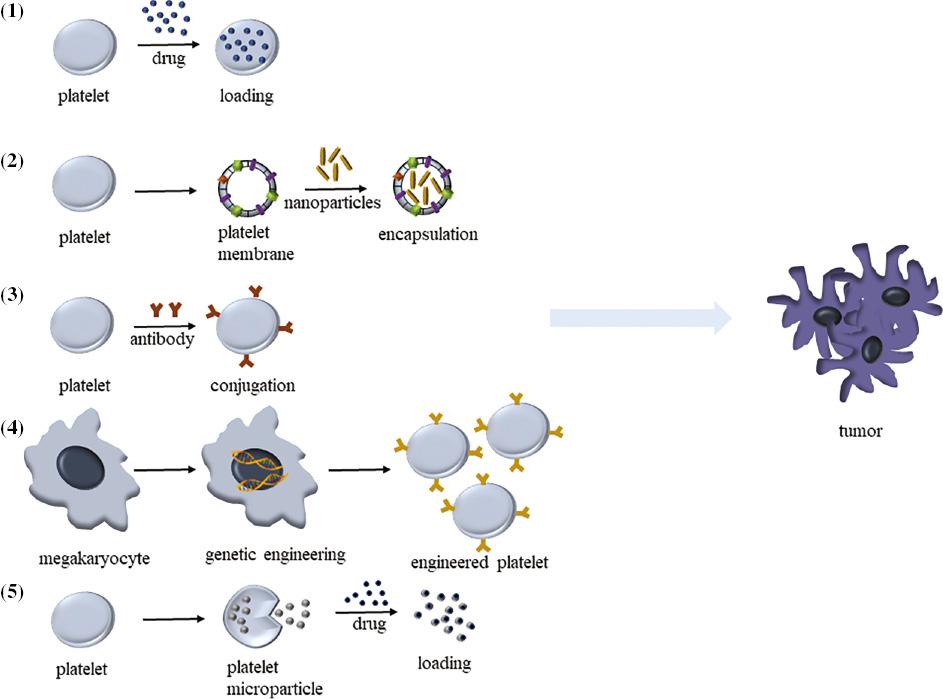
Schematic illustration of various platelet-based drug delivery systems.
(1) Platelets delivering antitumor drugs.
(2) Platelet membrane camouflaged antitumor drug delivery systems.
(3) Platelet engineering for drug delivery.
(4) Genetically engineered platelets deliver antitumor drugs.
(5) Platelet-derived microparticles for antitumor drug delivery.