
1. INTRODUCTION
Alzheimer’s disease (AD) is the most common neurodegenerative disease that progressively impairs memory and cognitive judgment [1]. AD severely affects the quality of life and imposes a heavy burden on the healthcare system [1]. The global incidence of AD currently exceeds 50 million, and the mortality rate due to AD has increased two-fold over the last two decades [2]. No cure for AD is currently available, and current therapeutics commonly used in the clinical setting only exert a modest effect in symptomatic relief, but fall short in reversing or even slowing down AD progression [3, 4].
The pathogenesis of AD is complex, much of which is unknown. There are two well-recognized pathologic hallmarks in AD (extracellular amyloid plaques and intracellular neurofibrillary tangles [NFTs]) [4]. The aggregation of beta-amyloid (Aβ) and hyperphosphorylated tau protein contribute to the formation of amyloid plaques ( Figure 1 ) and NFTs ( Figure 2 ), respectively, which are thought to have important roles in AD progression [4].
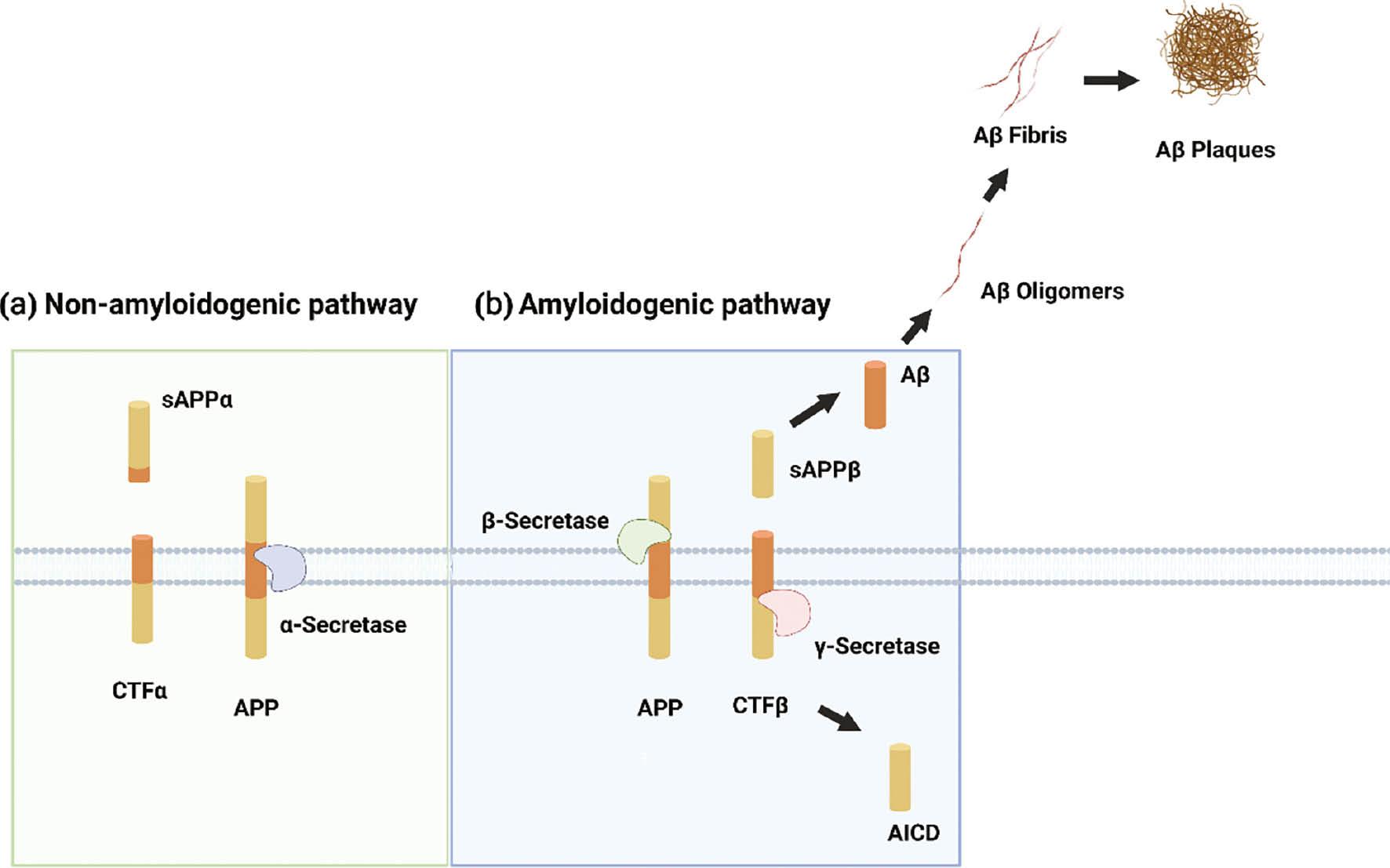
Amyloidogenic and non-amyloidogenic pathways for APP processing in AD.
There are two pathways of APP processing: (a) The non-amyloidogenic pathway, in which APP is cleaved by α-secretase, leads to the release of cytoplasmic tail fragment α (CTFα) and soluble APPα (sAPPα). sAPPα has neuroprotective effects due to prevention of APP degradation mediated by β-secretase. (b) The other pathway is the amyloidogenic pathway, in which β-secretase cleaves APP to produce CTFβ and soluble APPβ (sAPPβ), followed by γ-secretase-mediated cleavage to generate several types of Aβ. Aβ40 and Aβ42 are the two major Aβ peptides that have an important role in AD pathology. The ratio of these two isoforms is influenced by the pattern of cleavage from APP by α-, β-, and γ secretases. These two pathways are parallel and kept in balance under physiologic conditions. Dysfunction of Aβ clearance and degradation causes the deposition of Aβ in the AD brain, and sequentially generates Aβ oligomers and fibrils, ultimately leading to the formation of Aβ plaques.
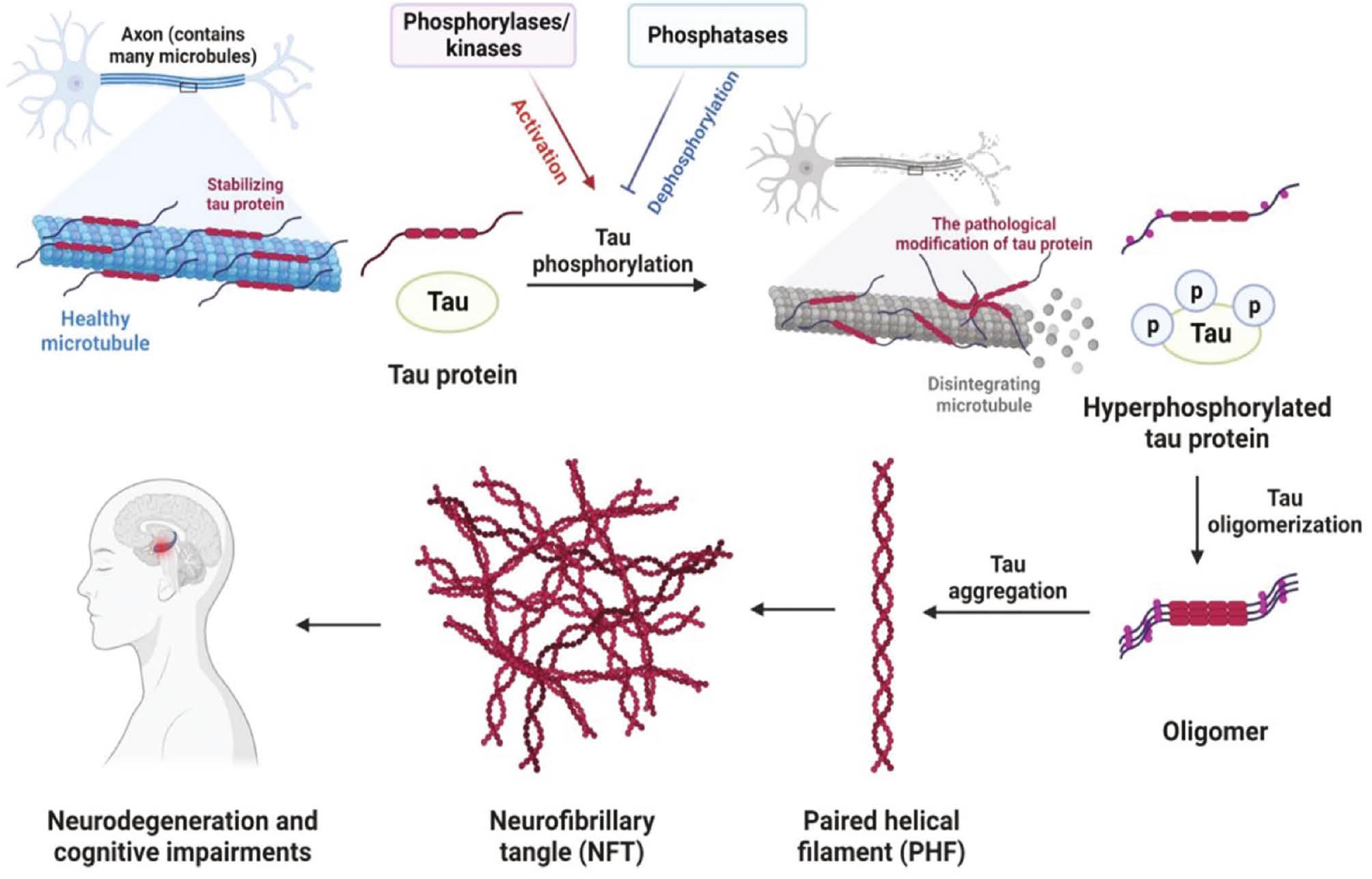
Intracellular neurofibrillary tangles (NFTs) pathway in AD.
Hyperphosphorylation occurs in an epitope-specific manner during the course of AD, and is known to underlie the mis-sorting of tau from a primarily axonal-to-a somato-dendritic location in neurons. Hyperphosphorylated tau loses its affinity to microtubules and destabilizes microtubule dynamics. After pathologic aggregation, abnormal folding of tau protein leads to the generation of a paired helical filament (PHF) and neurofibrillary tangles (NFTs), ultimately causing neurodegeneration and cognitive impairment in patients with AD.
The formation of amyloid plaques, mainly consisting of Aβ peptide, is one of the pathologic hallmarks in the brains of patients with AD. The amyloid cascade hypothesis proposes that Aβ accumulates and aggregates in the brain during the initial stage of AD pathology, and ultimately leads to neurodegeneration [5, 6]. Aβ is a 38-43-residue fragment formed by proteolysis of amyloid precursor protein (APP) through cleavage of β- and γ-secretases [7]. The following two pathways are associated with APP processing: the non-amyloidogenic pathway, in which APP is cleaved by α-secretase, leading to the release of cytoplasmic tail fragment α (CTFα) and soluble APPα (sAPPα) [8], which has neuroprotective effects due to its ability to prevent APP degradation mediated by β-secretase [9]; and the amyloidogenic pathway, through which β-secretase cleaves APP to produce CTFβ and soluble APPβ (sAPPβ), followed by γ-secretase-mediated generation of several types of Aβ [10]. Among the types of Aβ, Aβ40 and Aβ42 are the two major Aβ peptides that have important roles in AD pathology. The ratio of the Aβ40 and Aβ42 isoforms is influenced by the pattern of cleavage from APP by α, β, and γ secretases [11]. These two pathways are parallel and kept in balance in under physiologic conditions. Aβ clearance and degradation dysfunction can cause the deposition of Aβ in the brains of AD patients, and sequentially generate Aβ oligomers and fibrils, ultimately leading to the formation of Aβ plaques [6, 12]. In addition, the genetic mutations of the APP gene on chromosome 21, the presenilin-1 (PS1) gene on chromosome 14, and the presenilin-2 (PS2) gene on chromosome 1 are positively correlated with the increased levels of Aβ [13]. Accumulating evidence suggests that Aβ oligomers have significant neurotoxicity [14], and the Aβ plaques are usually surrounded by activated glial cells and damaged neurons [15–17]. It has been shown that soluble oligomeric and deposited Aβ in amyloid plaques interact with neurons, microglia, astroglia, and blood vessels, causing a variety of detrimental cellular responses that eventually lead to neuronal death [18].
NFTs are largely composed of hyperphosphorylated tau protein. NFTs are another pathologic hallmark of AD and contribute significantly to AD progression [19]. Tau protein is a glycoprotein related to microtubule assembly that has a crucial role in stabilizing microtubules, maintaining neuronal morphology, and modulating neurotransmitter transport under physical conditions [20]. Progression of AD is closely associated with abnormal post-translational modifications and aggregation of tau protein, including phosphorylation, acetylation, N-glycosylation, and truncation [21, 22]. Phosphorylation is the most common post-translational modification of tau protein. Accumulating evidence suggests that phosphorylated tau is closely correlated with AD symptom severity and may drive progression of AD [23]. In the brains of patients with AD, hyperphosphorylated tau protein loses affinity for microtubules and progressively forms insoluble intracellular NFTs, thus leading to neuronal dysfunction, synaptic loss, and ultimately cognitive impairment [24].
Tau and phosphorylated tau proteins have been reported to be associated with Aβ directly and indirectly. According to the amyloid cascade hypothesis, Aβ accumulation promotes the hyperphosphorylation of tau protein and the formation of NFTs [25]. Tau immunotherapy, however, alleviates amyloid pathology and memory impairment in a transgenic APP mouse model of AD [26], indicating that tau pathologies have important roles in Aβ-induced neurotoxicity. Multiple factors, such as calcium homeostasis, kinases (glycogen synthase kinase-3β [GSK-3β] and cyclin-dependent protein kinase-5 [CDK-5]), and free fatty acid disruptions, are responsible for the association between tau and Aβ pathologies. Moreover, tau and Aβ-associated pathologies exert synergistic effects on neurotoxicity and synaptic dysfunction [27]. The development of tau-targeted therapeutics may be a promising strategy for AD treatment, although the relationship between Aβ and tau has not been completely determined.
Other neuropathologic features, such as neuroinflammation, neuronal loss, oxidative stress, and synaptic impairment, are also believed to contribute to disease progression [28]. All of these mechanisms may interact, leading to amplification of the mechanisms and resulting in AD pathogenesis.
Animal models are important for investigating AD pathogenesis and evaluating potential therapies for AD. In the past few decades, many potential drug candidates have shown promising anti-AD effects in preclinical models, but surprisingly failed in clinical trials [29]. One of the reasons for this finding may be partially attributed to the inappropriate choice of preclinical models. Mammals, such as rodents, share extensive physiologic and genetic homology with humans. Thus, preclinical findings acquired from animal models may provide insight into human disease conditions. Although no single animal model can reproduce all aspects of the pathologic and behavioral features of AD patients, currently available AD animal models can, at least to some extent, replicate several important features of AD. This review aimed to summarize the characteristics and features of the animal models commonly in use for AD research. It is hoped that the information summarized in this article may help AD researchers choose appropriate animal models to achieve their objectives.
2. TRANSGENIC RODENT MODELS
Genetics is one of the most salient known risk factors for AD. AD can be divided into sporadic AD (sAD) and familial AD (fAD). The ε4 allele of the apolipoprotein E (ApoE) gene is considered the most relevant gene for sAD [30, 31], while fAD is mainly associated with familial gene mutations, including APP, PS1 and PS2 gene mutations [32, 33]. Compared with the most common ApoE genotype (ε3/ε3), ε4 heterozygosity raises the risk of developing AD 3-fold and ε4 homozygosity increases the risk 8-12 times [1]. Approximately two-thirds of pathology-confirmed AD cases are ε4-positive (homozygous or heterozygous), while the percentage of ε4-positive cases is 15%-20% in the general population [31]. Although pathogenic variants of APP, PS1, and PS2 genes contribute to ≤ 1% of AD cases [1], nearly 100% of individuals inheriting APP and PS1 and approximately 95% of individuals inheriting PS2 develop the disease, assuming an average lifespan [34].
The identification of APP, PS1, and PS2 gene mutations related to fAD [32, 33] has resulted in the development of numerous transgenic rodent models of amyloid pathology [35, 36], while the human MAPT gene mutations related to neurodegeneration [37] have been utilized to induce tau pathology. To date, > 200 transgenic rodent models of AD have been established (data obtained from https://www.alzforum.org/, accessed in November 2022), most of which are generated based on the mutations associated with amyloid and tau pathologies. Because ApoE4 and triggering receptor expressed on myeloid cells 2 (TREM2) genes are considered the most relevant genes for sAD [30, 31, 38], transgenic models associated with these genes have been established. The most commonly used transgenic rodent models of AD are listed in Table 1 .
Transgenic animal models used in the preclinical studies of AD.
| Model | Mutations | Genetic background and promoter | AD-related pathology | Behavioral impairment | Advantages | Disadvantages | References | ||
|---|---|---|---|---|---|---|---|---|---|
| Amyloid pathology | Tau pathology | Other neuropathologies | |||||||
| APP-based | |||||||||
| TgCRND8 | APPSwe, APPInd | Hybrid C3H/He-C57BL/6, hamster PrP promoter | Aβ deposition in cerebral cortex (2-3 months); hippocampus, midbrain, brainstem, and cerebellum with age | Hyperphosphorylated and aggregation (7-12 months) | 1. Gliosis (9-20 weeks) 2. Synaptic loss (6 months) 3. Cholinergic basal forebrain neurons (7 months) 4. Changes in LTP/LTD (6-12 months) | Learning and spatial memory deficits (3 months) | 1. Early-appearing Aβ deposition (2-3 months) 2. Hyperphosphorylated tau (7 months) 3. Early-appearing learning and spatial memory deficits (3 months) | No NFTs observed. | [39–43] |
| APP23 | APPSwe | C57BL/6, murine Thy1 promoter | Aβ deposition (6 months); plaques in cerebral cortex, hippocampus, thalamus, olfactory nucleus, and putamen (24 months) | Hyperphosphorylated tau in proximity to Aβ plaques; no NFTs observed | 1. Gliosis 2. Neuronal loss (14-18 months) | Learning and spatial memory deficits (3 months) and progress with age | Early-appearing learning and spatial memory deficits (3 months) | No NFTs observed | [44–47] |
| Tg2576 | APPSwe | C57BL/6×SJL, hamster prion protein promoter | Numerous parenchymal Aβ plaques (by 11-13 months). | None | 1. Gliosis (10-16 months) 2. Changes in LTP/LTD (5-15 months) | Impaired spatial learning, working memory, and contextual fear conditioning at < 6 months, although other studies have reported normal cognition at this age with progressive impairment by > 12 months | Early impaired spatial learning and memory functions (less than 6 months) | The absence of comprehensive tau pathology, including hyperphosphorylated tau and NFTs | [48–51] |
| PDAPP | APPInd | C57BL/6×DBA2, human PDGF-β promoter | Aβ deposition in hippocampus and cerebral cortex (6-9 months); extensive plaques with age | None | 1. Gliosis 2. Synaptic loss 3. Changes in LTP/LTD (4-5 months) | 1. Robust deficits (3 months) 2. Objective recognition deficits (6, 9-10 months) 3. Operative learning deficits (3-6 months) | Early changes in LTP/LTD (4-5 months) | The absence of comprehensive tau pathology, including hyperphosphorylated tau and NFTs | [52–56] |
| APP(V717I) | APPLon | Originally generated on FVB/N background; available at reMYND as C57BL/6×FVB/N, murine Thy1 promoter | Aβ deposition (2-4 months); plaque formation in hippocampus and cerebral cortex (13-18 months). | Hyperphosphorylated tau; no NFTs observed | 1. Gliosis (by 10 months) 2. Changes in LTP/LTD in hippocampus (6 months) | Learning and memory deficits (from 6 months) | 1. Early Aβ deposition (2-4 months) 2. Learning and memory deficits (from 6 months) 3. Changes in LTP/LTD in hippocampus (6 months) | No NFTs observed | [57, 58] |
| J20 | APPSwe, APPInd | C57BL/6, human PDGF-β promoter | Aβ deposition in cerebral cortex and hippocampus (5-6 months); progressive Aβ deposition with widespread plaques (by 8-10 months) | None | 1. Gliosis (24-36 weeks) 2. Synaptic loss (by 3 months) 3. Neuronal loss in CA1 region (12-36 weeks). 4. Changes in LTP/LTD (3-6 months) | Learning and spatial memory deficits (by 4 months) | 1. Early changes in LTP/LTD (3-6 months) 2. Early learning and spatial memory deficits (by 4 months) 3. Early synaptic loss (3 months) | The absence of comprehensive tau pathology, including hyperphosphorylated tau and NFTs | [59–62] |
| mThy1-hAPP751 | APPSwe, APPLon | C57BL/6×DBA, murine Thy1 promoter | Aβ deposition in frontal cortex (3-6 months); accumulation in cerebral cortex, hippocampus, thalamus, and cerebellum with age | None | 1. Gliosis (by 6 months) 2. Synaptic loss (from 12 months) | Learning and spatial memory deficits (by 6 months) | 1. Early appeared Aβ deposition in frontal cortex (3-6 months); 2. Early learning and spatial memory deficits (by 6 months); 3. Early gliosis (6 months) | The absence of comprehensive tau pathology, including hyperphosphorylated tau and NFTs | [63–65] |
| APPNL-G-F knock-in rats | APPSwe, APPArc, APPIbe | Sprague-Dawley, created using CRISPR/Cas9 technology | Amyloid plaques (1 month in homozygous knock-ins; 4 months in heterozygotes) | Hyperphosphorylated tau; no NFTs observed | 1. Gliosis (6 months in homozygous; 12 months in heterozygotes) 2. Synaptic loss 3. Neuronal loss (12 months) | Learning and spatial memory deficits (5-7 months) | 1. Early amyloid plaques (1 month in homozygous knock-ins; 4 months in heterozygotes) 2. Early learning and spatial memory deficits (5-7 months) | No NFTs observed | [66] |
| Tau-based | |||||||||
| Tau P301S | MAPT P301S | (C57BL/6 × C3H) F1, mouse prion protein promoter | None | NFTs in the hippocampus, neocortex, amygdala, brainstem, and spinal cord (6 months) with progressive accumulation | 1. Gliosis (by 6 months) 2. Synaptic loss (3-6 months) 3. Neuronal loss in hippocampus and cortex (9-12 months) 4. Changes in LTP/LTD (3-6 months) | 1. Learning and spatial memory deficits (6 months) 2. Impaired memory in assays of contextual fear conditioning | 1. Early synaptic loss (3-6 months) 2. Early changes in LTP/LTD (3-6 months) 3. Early learning and spatial memory deficits (6 months) 4. Early gliosis (by 6 months) | 1. The absence of amyloid pathology 2. No association between genetic mutation and tau pathology is found in AD patients | [67–69] |
| rTg(tauP301L)4510 | MAPT P301S | Mixed: 129S6 (activator)×FVB (responder), human CaMKIIα promoter | None | Pre-tangles (2.5 months); argyrophilic tangle-like inclusions in hippocampus (5.5 months) and cortex (4 months) | 1. Neuronal loss in hippocampus (from 5.5 months) 2. Synaptic loss (8-9 months) 3. Changes in LTP/LTD (4.5 months) | Learning and spatial memory deficits (2.5-4 months) | 1. Early pre-tangles (2.5 months) 2. Early learning and spatial memory deficits (2.5-4 months) 3. Early changes in LTP/LTD (4.5 months) 4. Early neuronal loss in hippocampus (from 5.5 months) | 1. The absence of amyloid pathology 2. No association between genetic mutation and tau pathology is found in AD patients. 3. The neuropathologic deficits and behavior phenotypes in this model may be partially attributed to its overexpression of mutant human tau | [70–74] |
| hTau | MAPT (all 6 isoforms) | The targeted allele was created in 129S4/SvJae-derived J1 embryonic stem cells that were subsequently injected into C57BL/6 blastocysts. The transgenic allele was generated in embryos derived from a cross between Swiss Webster and B6D2F1. Mice containing both alleles were back-crossed to C57BL/6 mice. | None | Hyperphosphorylated tau (from 6 months); aggregated tau and PHFs (9 months) | 1. Neuronal loss (8-18 months) 2. Changes in LTP/LTD (12 months) | 1. Learning and spatial memory deficits (6-12 months) 2. Impaired burrowing (4 months) | 1. Early impaired burrowing (4 months) 2. Early hyperphosphorylated tau (from 6 months) | The absence of amyloid pathology | [75–79] |
| THY-Tau22 | MAPT P301S, MAPT G272V | C57BL6/CBA; backcrossed to C57BL6. Thy1.2 promoter | None | Tau aggregation in cerebral cortex and hippocampus (3 months); hyperphosphorylated tau and NFTs in hippocampus and frontal cortex (6 months); age-related increase of tau pathology. | 1. Gliosis (age-dependent increase) 2. Neuronal loss in CA1 region of the hippocampus (from 12 months) 3. Changes in LTP/LTD (9-10 months) | 1. Spatial memory deficits (after 9 months) 2. Impaired appetitive responding | Early appeared Tau aggregation | 1. The absence of amyloid pathology 2. No association between genetic mutation and tau pathology is found in AD patients. | [80–83] |
| PSEN-based | |||||||||
| PS cDKO | PSEN1 conditional KO; PSEN2-/- | C57BL6/129 hybrid | Decreased Aβ deposition | Hyperphosphorylated tau (6 m). | 1. Gliosis (6 months) 2. Synaptic loss (6-9 months) 3. Neuronal loss (2-4 months) | 1. Learning and spatial memory deficits (mild at 2 months, age-dependent) 2. Contextual fear conditioning deficits (mild at 2 months, age-dependent) | 1. Early-appearing learning and spatial memory deficits, as well as contextual fear conditioning deficits (2 months) 2. Early-appearing neuronal loss (2-4 months) | This model cannot exhibit pronounced AD-like pathologies. | [84–86] |
| Multi-transgenic | |||||||||
| APP/PS1 | APPSwe, PSEN1 M146L | Tg2576×C3H, hamster prion protein promoter/PDGF-β promoter | Large amounts of Aβ accumulate in the cerebral cortex and hippocampus (from 6 months, increasing with age) | Hyperphosphorylated tau in proximity to Aβ deposits (from 6 months) | 1. Gliosis (6-12 months, increasing with age) 2. Neuronal loss in the CA1 region of the hippocampus (22 months) | Learning and spatial memory deficits (from 12-14 weeks) | 1. Early-appearing large amounts of Aβ accumulate (6 months) 2. Early-appearing hyperphosphorylated tau in proximity to Aβ deposits (from 6 months) | No NFTs observed. | [87–91] |
| PS2APP | APPSwe, PSEN2 N141I | C57BL/6, mouse prion protein promoter/Thy1.2 promoter | Aβ deposition (6 months); plaques in cerebral cortex and hippocampus (10 months) | None. | 1. Gliosis 2. Changes in LTP/LTD (10 months) | Learning and spatial memory deficits (8-12 months) | Early appeared Aβ deposition (6 months) | The absence of comprehensive tau pathology, including hyperphosphorylated tau and NFTs | [92–94] |
| 5×FAD | APPSwe, APPFlo, APPLon, PSEN1 M146 L, PSEN1 L286V | C57BL/6×SJL, mouse Thy1 promoter | Aβ plaques in deep cortical layers and subiculum (2 months); Aβ deposition in spinal cord (4 months); plaques throughout cerebral cortex and hippocampus (6 months). | None. | 1. Gliosis 2. Synaptic loss 3. Neuronal loss (12 months) 4. Changes in LTP/LTD | Learning and spatial memory deficits (3-6 months, age-dependent) | 1. Early-appearing Aβ plaques 2. Early-appearing learning and spatial memory deficits (3-6 months) | 1. The absence of comprehensive tau pathology, including hyperphosphorylated tau and NFTs. 2. Gender-dependently differences in phenotypes | [95–98] |
| 3×Tg | APPSwe, MAPT P301S, PSEN1 M146L | C7BL/6;129X1/SvJ;129S1/Sv, mouse Thy1.2 promoter | Aβ deposition in cerebral cortex (6-12 months); accumulation in cerebral cortex and hippocampus (12-20 months) | Hyperphosphorylated tau in hippocampus (12 months) and cerebral cortex (18 months) | 1. Gliosis (7 months) 2. Changes in LTP/LTD (6 months) | 1. A retention/retrieval deficit (by 4 months) 2. Learning and memory deficits (6.5 months) | 1. Early-appearing a retention/retrieval deficit (by 4 months) 2. Early-appearing learning and memory deficits (6.5 months) 3. Early-appearing changes in LTP/LTD (6 months) 4. Early-appearing Aβ deposition (6-12 months) | No association between genetic mutation and tau pathology is found in AD patients. | [99–101] |
| PLB1-triple | APPSwe, APPLon, MAPT P301 L, MAPT R406W, PSEN1 A246E | C57BL/6, mouse CaMKII-α promoter | Aβ deposition in cerebral cortex and hippocampus (5-6 months); plaque formation (12 months) | Hyperphosphorylated tau in the hippocampus and cortex (6 months). | Changes in LTP/LTD (6-12 months). | 1. Social recognition memory deficits (from 5 months) 2. Objective recognition memory deficits (by 8 months) 3. Spatial memory deficits (12 months) | 1. Early-appearing social recognition memory deficits (from 5 months) 2. Early-appearing Aβ deposition (5-6 months) 3. Early-appearing hyperphosphorylated tau in the hippocampus and cortex (6 months) | 1. The neurodegenerative pathologies progress slowly and are mild. 2. This model expresses low levels of mutant human APP, tau and PS1, with no NFTs and virtually absent Aβ deposition | [102–106] |
| E4FAD | APPSwe, APPFlo, APPLon, PSEN1 M146L (A>C), PSEN1 L286V, APOE Knock-in | C57BL/6 | Aβ plaques in the subiculum and deep cortical layers (by 4 months) | No data | 1. Gliosis (6 months) 2. Synaptic loss (by 4 months) | Modest learning and spatial memory deficits (by 2 months); worsen with age | 1. Early-appearing modest learning and spatial memory deficits (2 months) 2. Early-appearing Aβ plaques (4 months) 3. Early-appearing synaptic loss (by 4 months) | 1. The AD phenotypes are generally less severe than the original 5×FAD mouse model. 2. Tau pathology is not reported. | [107–109] |
| Trem2 R47H KI (Lamb/Landreth) ×APPPS1-21 | TREM2 R47H, APPSwe, PSEN1 L166P, Trem2 Knock-In | C57BL6/J, Thy1 promoter | Reduction in the number and burden of fibrillar amyloid plaques in the hippocampus | Hyperphosphorylated tau in dystrophic neurites surrounding plaques (4 months); no NFTs observed | Fewer plaque-associated myeloid cells | No data | Early-appearing hyperphosphorylated tau in dystrophic neurites surrounding plaques (4 months) | 1. No NFTs observed 2. Behavior deficits are not reported | [110–114] |
| PS19 with humanized TREM2 (R47H) | MAPT P301S, TREM2 R47H, Trem2 Knock-out | C57BL/6 | No data | Tangles revealed using antibody PG5 (9 months) | 1. Decreased astroglia and microglial reactivity (9 months) 2. More synapses and fewer dystrophic synapses (9 months) | No data | More synapses (9 months) | 1. No NFTs observed. 2. Amyloid pathology and behavior deficits are not reported | [59, 115, 116] |
Note: APPFlo, APP I716V; APPInd, APP V717F; APPLon, APP V717I; APPSwe, APP KM670/671NL.
2.1 APP-based transgenic model
The PDAPP transgenic mouse model, the first reported transgenic AD mouse model, was generated in the mid-1990s [52–56], followed by the development of the currently most commonly used APP transgenic models, including Tg2576 [117], APP23 [44–47], TgCRND8 [39–43], and J20 [118] transgenic mouse models. These five models overexpress human APP with the Indiana mutation (V717F) and/or the Swedish mutation (KM670/671NL). Other APP transgenic murine models, such as APP(V717I) and mThy1-hAPP751, encode a mutant form of human APP with the London mutation (V717I) driven by the murine Thy-1 promoter [57, 58, 63–65]. APP-based transgenic rodent models exhibit comprehensive Aβ pathology, Aβ-associated neuroinflammation, synaptic dysfunction, neuronal loss, and cognitive and behavioral impairment (also shown in Table 1 ), thus supporting the amyloid cascade hypothesis and recapitulating several key pathologies observed in AD patients. Indeed, these AD models are ideal experimental tools for amyloid-related research, not only for evaluating the potential efficacy of new therapeutics, but also investigating the underlying mechanisms, including amyloid pathology and amyloid-associated neuropathogenesis. It is worth noting, however, that the importance of Aβ in AD pathogenesis has been increasingly questioned due to the continuous clinical failures of anti-Aβ or amyloid-centric therapies [119–121]. Although aducanumab, a monoclonal antibody targeting Aβ pathology, was conditionally approved in 2021 by the United States Food and Drug Administration (US FDA) for the treatment of AD patients, although the clinical efficacy and safety have yet to be verified [122]. This therapeutic dilemma may be attributed to the complex pathologies of AD [123], while APP transgenic rodent models only display amyloid pathology. It is conceivable that the absence of comprehensive tau pathology in APP transgenic models, another crucial pathologic feature of AD, may result in the promising therapeutic effects of potential drug candidates demonstrated in animal models, but shown to be negative in AD patients. Although tau protein is hyperphosphorylated in some models, such as TgCRND8 [124], APP23 [44–47], and APP(V717I) mice [57, 58, 63–65], as well as APPNL-G-F knock-in rats [66], no NFTs were observed in APP-based transgenic rodent models. Therefore, the lack of NFT development in the above-mentioned amyloidosis models, at least to some extent, limits preclinical use [36, 125].
2.2 Tau-based transgenic models
Apart from amyloid plaques, another pathologic hallmark of AD is the formation of NFTs, which results from the hyperphosphorylation and pathologic aggregation of tau proteins [19]. Because the formation of NFTs is absent in transgenic rodents harboring APP or APP/PS mutations [126], mutated MAPT models are often selected to investigate tau pathology in preclinical studies on AD.
2.2.1 hTau.P310S
As the most widely used tauopathy model, tau P310S mice express mutant human tau 5-fold higher than the endogenous murine tau on a C57BL/6×C3H background, and display widespread NFTs in the hippocampus, neocortex, amygdala, brainstem, and spinal cord at 6 months of age, with progressive hippocampal synaptic dysfunction at 3-6 months of age and neuronal loss by 9-12 months of age [67]. Tau pathology is also accompanied by microglial and astrocytic activation by 6 months of age [67]. Moreover, 6-month-old P301S transgenic mice exhibit learning and spatial memory deficits, when undergoing the Morris water maze test (MWMT) and impaired spontaneous alternating behavior in the Y-maze test [68].
2.2.2 rTg (tauP301L) 4510
After the initial description in 2005 [70, 71], the rTg4510 mouse model has gradually become a popular and commonly used tauopathic model. rTg4510 mice encode a repressible mutant of human tau with a P301L mutation, expressing mutant human tau at a level 13 times higher than endogenous murine tau [71]. NFT-like pathologic changes appear in the cortex and hippocampus at 4 months of age and 5.5 months of age, respectively [70, 71]. Apart from an early onset of tau pathology, this model shows progressive and age-related spatial memory impairment beginning at 2.5 months of age [71, 72], while females exhibit more severe deterioration than males [72]. Hyperactivity and recognition memory deficits are also observed, as demonstrated by the open field and novel objective recognition tests [127–129]; however, a recent study suggested that the neuropathologic deficits and behavior phenotypes in this model may be partially attributed to overexpression of mutant human tau [130]. Thus, researchers are advised to take factors other than hTau overexpression into consideration when using rTg4510 mice.
2.2.3 hTau
Unlike the above-mentioned animal models of tau pathology, hTau mice develop tangles without mutations and exclusively express all six isoforms of non-mutant human tau, including 3R and 4R forms, but no endogenous mouse tau is detected [75]. Tau protein is progressively hyperphosphorylated and accumulated from 6 months of age, while paired helical filaments (PHFs) and thioflavin-S-positive NFTs can be detected in 9-15-month-old hTau mice [75]. Compared with the wild-type littermates, hTau mice exhibit significant deficits in spontaneous burrowing behavior at 4 months of age [76], as well as spatial learning and memory at 6-12 months of age [77, 78], with neuronal loss detected at 8-18 months of age [79].
2.2.4 THY-Tau22
This model encodes a transgene containing human tau with P301S and G272V mutations driven by a Thy1.2 promotor. The tau pathologies with an onset at 3-6 months develop progressively with age in THY-Tau22 mice, including hyperphosphorylation of tau protein and the formation of NFTs in the hippocampus and cortex [80]. In addition, this model shows neuronal loss, synaptic dysfunction, and an age-dependent increase in GFAP-positive astrocytes in the hippocampus [80, 81, 131]. THY-Tau22 mice exhibit spatial memory impairment at 9 months of age [81] and impaired appetitive responding at 9-10 months [82].
Although tau-based rodent models display pronounced tau pathology, Aβ plaques and amyloid pathology are absent in these models [70, 71, 75, 80, 132]. Of note, pathogenic mutations of the tau gene are only present in patients with frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), but not in individuals affected with AD [133]. Due to the lack of amyloid pathology in tau-based models and the lack of tau pathology in APP-based models, these AD models cannot recapitulate the association between tau and amyloid pathologies observed in AD patients. Thus, some multi-transgenic AD models, such as triple transgenic (3 × Tg) and PLB1-triple mice, have been generated to counterbalance the above-mentioned shortcomings.
2.3 PSEN-based transgenic models
As major components of the γ-secretase enzyme, PS1 and PS2 are closely associated with APP processing and AD pathology [134, 135]. Mutations of these two genes contribute to the early onset of fAD, in which PS1 mutations play a more prominent role [134, 135]. Transgenic models only expressing mutant PS1 or PS2 produce a significantly elevated level of Aβ42, a major Aβ peptide preceding Aβ plaque deposition, but Aβ plaques and NFTs are not developed [136]. In addition, age-related tau inclusions, synaptic dysfunction, and neuronal loss are present in transgenic animals expressing mutant PS1 or PS2 [137–139]. These models generally do not exhibit pronounced AD-like pathologies because the models have a lower Aβ42 level than APP transgenic mice [136, 140]. Thus, the application of PS mutant mice is limited; however, the development of APP/PS transgenic mice has drawn much attention because the PS1 or PS2 mutations have synergistic effects with APP on accelerating amyloidosis [136].
2.4 Multi-transgenic models
2.4.1 APP/PS1
Compared to single APP transgenic models, APP/PS mice have a more extensive and earlier onset of amyloid pathologies and cognitive deficits, in addition to more pronounced synaptic and neuronal loss [136, 137]. Specifically, APP/PS1 mice harboring mutant APPswe and PS1 (M146L) display Aβ depositions much earlier than the original monogenic APP transgenic mice (Tg2576 mice) [141]. Other APP/PS1 lines, such as APPSwe × PS1△E9, APPSwe × PS1A246E, and APPLon × PS1A246E, have also been developed and widely used. The extensive applications of APP/PS1 mice in preclinical AD research are mainly attributed to rapid and age-dependent Aβ accumulation and deposition in the hippocampus and cortex at an early age, with an enhanced Aβ42/Aβ40 ratio and behavioral impairments. Like monogenic APP transgenic mice, however, tau pathologies are absent in APP/PS1 mice.
2.4.2 5×FAD
The 5×FAD model is used commonly in preclinical AD-associated research. These mice harbor five AD-related human mutant genes: mutant APP (with Swedish, Florida, and London mutations); and PS1 (with M146 L and L286V mutations). Like other APP/PS1 mice, this model has a rapid and early onset of amyloid deposition, making APP/PS1 mice a valuable preclinical tool for amyloid-associated studies. Extracellular Aβ plaques appear in the hippocampus and cortex beginning at 2 months of age and progress in an age-dependent manner, while accompanied with neuroinflammation [95–98]. Of note, female mice generate higher Aβ levels than males and display more numerous Aβ plaques in the hippocampus and cortex, showing more aggressive amyloid pathology [142–144]. This gender-dependent difference in phenotypes is generally similar to AD patients, as women have been demonstrated to be more susceptible to AD than men [1]. Moreover, 5×FAD mice exhibit age-dependent cognitive deficits, including impaired spatial memory and working memory [95–98], with neuronal loss and synaptic dysfunction [95–98, 145].
2.4.3 3×Tg-AD
As the most commonly used transgenic AD mouse model, 3×Tg-AD mice encode three mutant forms of genes associated with fAD (APPSwe , MAPT P301L, and PSEN1 M146V) and develop both amyloid and tau pathologies [99]. Extracellular Aβ deposition occurs in the cerebral cortex by 6 months of age and becomes more extensive by 12 months, while tau protein is hyperphosphorylated and aggregated in the hippocampus by 12-15 months of age, which is later than amyloid deposition [99, 100]. Neuroinflammation and synaptic deficits appear at 7 and 6 months of age, respectively; both neuroinflammation and synaptic deficits occur prior to amyloid and tau pathologies [99, 146]. These mice show an early onset of intraneuronal Aβ-associated cognitive impairment at 4 months of age that manifests as a deficit with long-term retention [100]. Thus, this model recapitulates many AD-associated phenotypes and is well-recognized as one of the most appropriate models for mechanistic investigations and drug discovery in AD.
2.4.4 PLB1-triple
The PLB1-triple mice have five transgenic mutations on a C57BL/6J background, including APP Swe, APP Lon, MAPT P301 L, MAPT R406W, and PSEN1 A246E. Unlike models with high overexpression of disease genes, this model expresses low levels of mutant human APP, tau, and PS1, with no NFTs and virtually no Aβ deposition [102]. The neurodegenerative pathologies in this model progress slowly and are mild; however, spatial memory deficits are reported in 12-month-old PLB1-triple mice, with sparse Aβ plaques and hyperphosphorylated tau in the hippocampus and cortex from 6 months onwards [102, 103].
2.4.5 E4FAD
As mentioned above, the APOE4 gene is currently the most relevant gene associated with the development of sAD [30, 31]. The APOE4-elicited increase in the risk of developing AD has been reported to be closely related to the high avidity and specific binding of Aβ peptide with APOE4, sequentially leading to enhanced Aβ aggregation and reduced Aβ clearance [30, 31, 147]. Thus, some animal models have been established to investigate the role of APOE4 in AD, such as the E4FAD mouse model. The E4FAD mice are generated through cross-breeding of 5×FAD mice with the APOE4-targeted replacement mice on a C57BL/6 background. In this model, the AD phenotypes are generally less severe than the original 5×FAD mouse model. While Aβ plaques can be detected in 5×FAD mice at 2 months of age, the E4FAD mouse model exhibits progressive amyloid plaques in the cerebral cortex at 4 months [107]. The E4FAD mice have increased compact plaques and decreased diffuse plaques in the brain relative to mice with other APOE isoforms, such as E2FAD mice expressing APOE2 and E3FAD mice expressing APOE3 [107]. Moreover, age-dependent cognitive impairments in MWMT and Y maze are found in this model at 2 months, with elevated neuroinflammation at 6 months and synaptic loss at 4 months [108, 109] compared to the E2FAD and E3FAD models.
3. NON-TRANSGENIC ANIMAL MODELS
Most transgenic animal models are generated based on genetic mutations, bringing valuable insight into the roles of amyloid and tau pathogenesis in AD; however, > 95% of AD patients are diagnosed with sAD and are rarely caused by familial gene mutations [148, 149]. Although the etiology of sAD has yet to be fully elucidated, some non-transgenic animal models have been established to mimic the neuropathologic changes in sAD. The most commonly used non-transgenic animal models of AD are shown in Table 2 .
Non-transgenic animal models used in the preclinical studies of AD.
| Model | AD-related pathophysiology | Behavioral impairment | Mechanisms | Advantages | Disadvantages | References |
|---|---|---|---|---|---|---|
| Natural aging-based AD-like models | 1. Cholinergic hypofunction and other neurotransmitter alterations 2. Morphologic alterations | Learning and memory deficits (mice older than 18 months, rats older than 24 months and monkeys older than 16 years) | Aging | 1. Increased knowledge of neural mechanisms of cognitive deficits 2. Defining normal ageing versus pathological states | 1. Long experimental cycle 2. High maintenance cost | [150] |
| Senescence-accelerated mouse (SAM) models | 1. Hyperphosphorylated tau, Aβ deposition, and neurotransmitter dysfunctions in SAMP8 mice 2. Aβ deposition, neuronal loss, and neuroinflammation in SAMP10 mice | Learning and memory deficits (after 6 months for SAMP8 mice and 4-6 months for SAMP10 mice) | Aging | 1. Increased knowledge of neural mechanisms of cognitive deficits 2. Defining normal ageing versus pathologic states | 1. Poor drug absorption. 2. High mortality rate 3. The pathologies in these models are inconsistent with the pathogenesis of AD | [36, 151–157] |
| Streptozotocin (STZ)-induced AD model | 1. Hyperphosphorylation tau in the hippocampus and cortex (3 weeks) 2. Increased Aβ40 and Aβ42 levels; increased BACE1 expression; Aβ deposition 3. Insulin deficiency 4. Activated glial cells and increased pro-inflammatory cytokines (after 7 days) 5. Neuronal loss (after 2 weeks) 6. Oxidative stress | Learning and spatial memory deficits (after 2 weeks) | Insulin signaling pathway/neuroinflammation | 1. Increased insight into roles of specific pathophysiologic pathways in neurodegeneration 2. Preclinical evaluation of drugs targeting a specific pathway, such as the insulin signaling pathway | 1. The absence of obvious NFTs 2. Repeated or prolonged exposure to STZ is a health hazard and possibly carcinogenic to humans 3. The STZ solution is unstable and has a short half-life 4. High resistance to STZ injection in the female animals | [158–167] |
| D-galactose-induced AD animal model | 1. Oxidative stress (8 weeks) 2. Activated microglia and astrocyte (after 6 weeks) 3. Abnormal acetylcholine levels and choline acetyltransferase activity | Spatial memory impairment (after 6 weeks) | Oxidative stress | This model is applicable to study the AD pathogenesis in aging | 1. No development of NFTs 2. No Aβ pathology 3. Gender-dependent differences in pathologic changes | [168–173] |
| Aluminum-induced rodent model | 1. Hyperphosphorylated tau 2. Overexpression of the APP gene (4 months); increased Aβ42 level and BACE1 expression (6 weeks) 3. Oxidative stress; increased MDA level, decreased SOD activity, and DNA oxidative damage in the hippocampus and cortex (60-100 days) | Learning and memory deficits (after 6 weeks) | Metal ions disorder | Preclinical evaluation of the relationship between metal ion dysfunction and AD pathology. | The absence of Aβ plaques and NFTs | [174–183] |
| Scopolamine-induced model | Cholinergic hypofunction | Cognitive deficits | Brain atrophy | 1. Increased insight into the role of the cholinergic system in cognition 2. Preclinical evaluation of cholinomimetics | 1. No pathologic AD hallmarks 2. No disease progression | [184–189] |
| Aβ infusion (single or chronic) | Increased Aβ levels (after 2 weeks) | Learning and memory deficits (after 2 weeks) | Aβ deposits | 1. Increased insight into Aβ toxicity 2. Preclinical evaluation of Aβ targeting drugs | 1. No development of NFTs 2. Hyper-physiologic Aβ levels required | [190–197] |
| Acrolein-induced model | 1. Hyperphosphorylated tau 2. Increased secretion of Aβ; increased BACE1 expression 3. Increased MDA level | Learning and memory deficits | Oxidative stress | NA | No typical Aβ plaques and NFTs observed | [183] |
| Traumatic brain injury (TBI)-induced models | 1. Aβ deposition 2. Abnormal tau aggregation and hyperphosphorylation 3. Activated glial cells and increased pro-inflammatory cytokines 4. BBB dysfunctions 5. Neuronal loss | Learning and spatial memory deficits | TBI-induced phosphorylation of tau protein | This model is applicable for studies on the regulatory mechanism of hyperphosphorylation of tau protein. | 1. No obvious Aβ plaques and NFTs 2. Poor comparability among different TBI models due to the absence of officially neurologic criteria for trauma | [198] |
| High cholesterol diet-induced AD animal model | 1. Increased Aβ levels (after 3 weeks); increased APP expression (after 2 weeks) 2. Activated microglia and increased pro-inflammatory cytokines (after 1 week) 3. Oxidative stress 4. BBB dysfunction | None. | Hypocholesteremia | This model is applicable to study the relationship between lipid metabolism disorders and AD pathologic progress | 1. Seldom occurrence of tau hyperphosphorylation 2. No behavior impairments observed | [199–202] |
3.1 Spontaneous models
3.1.1 Senescence-accelerated mouse (SAM) models
The most salient known risk factors for AD are the non-modifiable contributors of older age, gender, genetics, and family history. Of these risk factors, advanced age has the most significant known impact on the risk of developing AD [1]. Thus, some AD models are established based on aging. The SAM, a mouse model of AD established in the early 1980s through phenotypic selection from a genetic pool of AKR/J mice, has mostly duplicated natural age-associated deterioration [36]. The SAM model consists of nine major sub-strains of SAM-prone (SAMP) mice, including the SAMP8 and SAMP10 sub-strains. Accumulating evidence demonstrated that SAMP8 mice display progressive memory deficits from 6 months of age and other aging features at a young age, such as sparse hair and osteoporosis [151, 152], with age-related hyperphosphorylated tau, Aβ deposition, and neurotransmitter dysfunction [153–155]. Therefore, the SAMP8 mouse model has been used as an aging-associated AD model. Another sub-strain, the SAMP10 strain, has also been proven to be a relevant model for AD, as these mice show age-associated learning and memory deficits at 4-6 months of age and exhibit Aβ deposition, neuronal loss and neuroinflammation during the aging process [156, 157]. Due to the high mortality rate and poor drug absorption, the use of SAMP models is limited. Moreover, the pathologic alterations in these models are not consistent with AD pathogenesis [33].
3.1.2 Natural aging models
Natural aging animals have also been used for AD research. Although natural aging rodents show age-related cognitive impairments morphologic changes, and cholinergic hypofunction [150], aging rodents do not develop amyloid and tau pathologies, making them less useful for AD studies targeting these two pathologic features. Furthermore, the high cost of maintenance and long experimental cycle largely restrict preclinical use. Some other species, including dogs, goats, cats, polar bears, sheep, wolverines, and several non-human primate species, can spontaneously develop Aβ plaque pathology, and some species even exhibit tau pathology [36]. These histopathologic changes can be accompanied by learning and memory impairment; however, there are some shortcomings or obstacles when using these species for experimental studies, such as limited availability, ethical problems, difficulty of large-scale breeding, and the high maintenance cost.
3.2 Streptozotocin (STZ)-induced AD model
As a natural alkylating anti-neoplastic agent isolated from Streptomyces achromogenes, STZ is selectively toxic for insulin-producing cells in the brain and periphery. The administration of STZ via intracerebroventricular (ICV) injection progressively and in a dose-dependent fashion causes AD-like pathologies, including cerebral glucose/energy metabolism impairments, decreased brain insulin resistance state, Aβ plaque deposition, hyperphosphorylation of tau protein, synaptic dysfunction, neuroinflammation, oxidative stress, and neuronal loss [158–161]. Moreover, behavioral deficits, such as progressive memory impairment, anxiety- and depression-like behaviors, and social dysfunction, can also be induced by ICV injection of STZ in rodents [162–164]. These pathologic alterations are generally consistent with the features of sAD patients. Thus, STZ has been extensively used to establish the sAD rodent model for the mechanistic assessment and preclinical evaluation of potential therapeutics.
Sone features or disadvantages in this model can hinder preclinical use. First, the STZ-induced AD model does not develop pronounced NFTs, although tau protein is hyperphosphorylated in the hippocampus and cortex 3 weeks after injection [165]. Second, repeated or prolonged exposure to STZ is considered a health hazard and possibly carcinogenic to humans [166]. Therefore, qualified experimental laboratories and appropriate personal protective equipment are required during the establishment of the STZ-induced AD model. Third, the STZ solution is unstable and has a short half-life (15-30 min), thereby easily leading to failure of model establishment if not handled properly. In addition, females are more resistant to pathologic changes induced by STZ injection than male rats, thus there are significant sex differences in cognitive impairment, glucose uptake, glutathione level, hippocampal astrocyte activation, and the choline acetyltransferase level [167]. The sex-dependent phenotypes in this model are inconsistent with the clinical findings that showed women have a nearly two-fold increased risk of developing AD than males [1].
3.3 D-galactose-induced AD model
D-galactose (D-gal) is a naturally-occurring monosaccharide sugar that serves as an energy-providing source and essential glycosylation component in mammals. As a reducing sugar, D-gal can be metabolized in the body under physiologic conditions, while high levels of D-gal can be changed to aldose and hydroperoxide under catalysis of galactose oxidase [168, 169]. The increased production of reactive oxygen species (ROS) induced by high D-gal levels cause oxidative stress, ultimately accelerating the process of aging [168, 170]. Accumulating evidence has shown that rodents receiving a continuous subcutaneous or intraperitoneal injection of D-gal exhibit progressive learning and memory impairment, accompanied by mitochondrial dysfunction, oxidative stress, microglial and astrocytic activation, and impaired cholinergic neurons in the brain [169, 171, 172]. All of these pathologic alterations are generally consistent with the natural brain aging process. Thus, long-term injection of rodents with D-gal has been widely used to study the mechanisms involved with aging and age-associated neurodegeneration and evaluate potential therapeutic strategies. Because increasing age is the most salient known risk factor for developing AD [1], the D-gal-induced model is applicable to the research on AD pathogenesis in aging.
The D-gal-induced model does not develop NFTs and amyloid plaques, the two neuropathologic hallmarks in the AD brain, or tau and Aβ pathologies [169, 171]. Moreover, gender-dependent differences have been shown with respect to the pathologic changes induced by D-gal [173]. Chronic injection of 100 mg/kg D-gal for 6 consecutive weeks causes spatial memory deficits and cerebral oxidative stress in male mice, but not female mice [173], indicating that male mice are more vulnerable to D-gal-elicited oxidative damage.
3.4 Aluminum-induced rodent model
As the most abundant neurotoxic metal, aluminum (Al) is closely associated with AD pathogenesis [203, 204]. Mounting evidence demonstrates that Al can cross the blood-brain barrier (BBB) and accumulate in the brain in a semi-permanent manner [205, 206]. The high consumption of Al from drinking water has been reported to increase both the incidence of cases and AD mortality [207, 208], and Al has been shown to promote the neurofibrillary degeneration and accelerate the formation of NFT-like structures in AD patients [209–211], suggesting that Al is a potential causative factor for AD. The association between Al exposure and tau pathology was later confirmed by experimental findings. Oshima et al. [174] reported that chronic oral administration of Al accelerates tau aggregation, promotes apoptosis, and exacerbates motor dysfunction in a tauopathy mouse model, showing that Al has an aggravating effect on tau pathologies. Moreover, Al administration induce taus or tau-related pathologies in experimental animals by suppressing the dephosphorylation of tau protein, increasing tau aggregation, and enhancing the activities of GSK-3β and CDK5, the two important tau kinases [204]. Apart from the development of tau pathologies, Al exposure affects Aβ by increasing the aggregation and Aβ-associated endothelial cell toxicity [175, 176]. The APP gene has been reported to be overexpressed in rat brain after treatment with 1.7 mg/kg aluminum chloride (AlCl3) for 4 consecutive months [177], while the oral administration of 17 mg/kg AlCl3 for 6 consecutive weeks causes increased BACE-1 and Aβ42 levels in rats [178]. Other prominent pathologic features, such as oxidative damage [179], neuroinflammation [180, 204], neuronal apoptosis [181], and increased AChE activity [182], are also observed in an Al-treated AD model. All these neurologic alternations induced by Al exposure are attributable to the impaired cognitive functions in animals, which are generally congruent with that which is observed in the early stage of AD [203, 204]. Although Al causes neuropathologic and neurobehavioral changes in in vivo experimental models, poor bioavailability hinders use of Al due to low solubility at a physiologic pH. Thus, AlCl3 rather than Al is often used to establish the animal model of AD; however, it is worth noting that an aqueous solution of AlCl3 partially hydrolyzes and releases volatile hydrogen chloride [183], affecting its bioavailability. Furthermore, this Al-induced AD model does not develop Aβ plaques and NFTs, two typical hallmarks of AD.
3.5 Scopolamine-induced rodent model
Cholinergic neurons are extensively distributed in the central nervous system (CNS) and have a predominant role in regulating critical physiologic processes, such as attention, learning, memory, and the stress response [212]. dysfunction of the cholinergic transmission system contributes to AD pathology [213]. Cholinergic antagonists impair learning and memory, while the administration of cholinergic agonists increases the availability of acetylcholine (ACh) at synapses and ameliorates cognitive deficits [198, 213]. Scopolamine, a known non-selective antagonist of the postsynaptic muscarinic receptor, disrupts the cholinergic tracts for cognition and induces learning and memory impairment in animals via intraperitoneal or ICV injection [184–186]. Animals treated with scopolamine partially exhibit the pathologic features of patients with AD; specifically, the Ach level is decreased, cholinergic neuron loss, amnesia, oxidative damage, and neuroinflammation [187–189]. Thus, the scopolamine-induced animal model provides an experimental tool to investigate the role of the cholinergic system in cognition and assess the preclinical efficacy of cholinomimetics; however, this model lacks typical AD pathologic hallmarks and disease progression, imposing limitations on preclinical use.
3.6 Aβ infusion (single or chronic)
The amyloid cascade hypothesis proposes that cerebral accumulation and aggregation of Aβ in the initial stage of AD is the primary culprit driving pathogenesis, regardless of sporadic or familiar AD, which ultimately results in neurodegeneration [5, 6, 12]. The Aβ-associated pathologies of the AD brain can be mimicked by intrahippocampal, intracerebral, or ICV injection of different Aβ species into the rodent brain [214]. Aβ peptides, including Aβ40, Aβ42, and Aβ25-35, can be administered in single or multiple injections to induce AD-like pathologies and behavioral deficits [190–192], including cholinergic dysfunction, Aβ deposition, and learning and memory impairment [191, 193–195]. In addition, the severity of cognitive impairment is associated with the species of Aβ infused and the interval between Aβ administration and behavioral tests.
In addition to the APP transgenic animal models, this Aβ infusion rodent model provides researchers with alternative means to study the mechanisms underlying Aβ toxicity in vivo and assess the preclinical effects of Aβ-targeting drugs. Moreover, this model has a much shorter experimental cycle than transgenic models. As alluded to above, it takes several months to observe Aβ plaque deposition in APP-based transgenic rodents, while plaque deposition can be found within a few weeks after Aβ infusion in animals [196]; however, like APP transgenic models, the formation of NFTs are not observed in this Aβ infusion model, suggesting that Aβ infusion does not induce essential neuropathologic features of AD. Furthermore, hyperphysiologic Aβ levels required to establish this model are much higher than the Aβ concentrations in the cerebrospinal fluid or brains of AD patients [197].
3.7 Other non-transgenic AD models
In addition to the spontaneous and chemical/drug-induced models described above, AD-like pathologies can also be induced by acrolein [215] and traumatic brain injury [216] ( Table 2 ).
Moreover, in addition to rodents, other non-rodent animal species have also been used as AD animal models, such as Caenorhabditis elegans [217], zebrafish [218], Drosophila melanogaster (also known as fruit flies) [219], and non-human primates [215, 220]. These species, however, are much less used than rodents in AD studies due to the limited availability, the vast phenotypic or pathologic differences from humans, difficult large-scale breeding, ethical problems, and the high maintenance cost. In addition, C. elegans and D. melanogaster are valuable and advantageous AD models for genetic manipulation and high-throughput drug screening due to the short lifespan, ease of maintenance, and genetic tractability [221, 222]. These two models can be genetically modified to overexpress human APP and MAPT genes, and are useful for studying the amyloid and tau pathologies in AD [217, 219]. C. elegans has a short life cycle of 3 days from egg-to-adult at 25 °C [222], while D. melanogaster has a short lifespan that includes four different morphologic stages (embryo, larva, pupa, and adult) [223], each of which allows genetic dissection of the mechanisms that affect aging and lifespan. Thus, although higher animals (e.g., rodents and non-human primates) are necessary for a more comprehensive understanding of AD pathology and for developing and translating potential therapeutics, lower species, such as C. elegans and D. melanogaster, can replace higher species for specific studies.
4. CHALLENGES AND PERSPECTIVES
Currently, numerous animal models are available for AD research, all of which have their own advantages and disadvantages. Of note, no experimental model can perfectly replicate all aspects of the neuropathologic and behavioral features that occur in AD patients. For example, the absence of Aβ plaques is commonly noted in in tau-based transgenic models, and the lack of NFTs formation is a feature of most sAD models and APP-based transgenic models. Thus, the experimental findings from animal studies may not be directly translated into clinical use. Moreover, because of the dramatic increase in the incidence of AD and the absence of a promising cure for AD at present, some potential drugs have been evaluated in clinical trials without reliable and robust preclinical data from animal studies, hence heightening the likelihood of failure in a clinical study. To validate a successful translation, the potential therapeutic strategies or drug candidates should be evaluated in different AD models to test efficacy and safety because pathologic differences are detected in different AD animal models. In addition, to avoid the preclinical inconsistency among different researchers or laboratories, it is necessary to follow standard procedures or protocols to assure the results are comparable and repeatable. Some guidelines, such as the Planning Research and Experimental Procedures on Animals: Recommendations for Excellence (PREPARE) [224] and the Animal Research: Reporting of In Vivo Experiments (ARRIVE) [225] guidelines, should be considered for improving the methodologic quality during the design, execution, and reporting of further animal studies in AD.
Some key points should also be considered when using AD animal models. First, like AD patients, gender often affects not only the onset but also the progression of the disease in AD models, as reviewed above. In some AD models, females display earlier AD phenotypic and pathologic characteristics than males, such as 5×FAD transgenic mice. In contrast, males are more vulnerable to AD and exhibit more severe deterioration in other models, such as the STZ-induced AD animal model. Second, because most AD-like pathologies and cognitive deficits in animal models are time- or age-dependent, the appropriate timing of behavioral tests and tissue/sample collection should be designed with an intention to obtain better and more promising results. Third, although animal models can mimic AD-like phenotypes, interspecies variability can cause marked pathologic differences, and the pharmacokinetics are potentially different between species [226]. Nevertheless, up to now, the use of AD animal models for preclinical research is well-recognized and well-accepted because the phenotypic similarities between humans and animals are more prominent than the differences [227].
5. CONCLUSIONS
This review comprehensively summarized the AD animal models and the model characteristics, including transgenic animal models, chemical- or drug-induced animal models, and spontaneous animal models. Some phenotypic and neuropathological changes in AD patients can be recapitulated with genetic and pharmacologic manipulations in animals. Despite the challenges and constraints, the currently available AD models remain very valuable tools for deciphering the pathogenic mechanisms underlying AD and developing new therapeutic strategies.