
1. INTRODUCTION
Protein homeostasis plays a key role in cell proliferation, differentiation, and cell death. As a major pathway for protein degradation, the ubiquitin-proteasome system (UPS) degrades target proteins through the action of ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin-protein ligases (E3). A new drug discovery strategy, proteolysis targeting chimeras (PROTACs), has attracted increasing attention in recent years [1]. PROTACs is typically composed of a protein-targeting ligand that is covalently linked to an E3 ligase ligand via an appropriate linker ( Figure 1 ) [2]. Through specific binding to the target proteins, the PROTAC recruits an E3 ligase and causes the ubiquitination and subsequent degradation of the target proteins via the UPS [3–5]. This technology has two advantages: the PROTAC-binding sites on the same target protein may be infinite [6], and binding and degradation activities may occur in multiple target proteins, thus enabling smaller drug dosages and pharmacological effect observations [7–9].

Since the first PROTAC molecule was developed by Crews, more than 50 target proteins have been successfully degraded ( Figure 2 ) [1, 10]. The PROTAC ARV-110 degrader has entered clinical trials (NCT03888612, Arvinas) for the treatment of metastatic castration-resistant prostate cancer via targeting the androgen receptor (AR) [2, 11].

A key consideration in the development of PROTAC drugs is selecting an appropriate combination of ligands for the E3 ligase and the target protein [12, 13]. In this context, the use of computational strategies to understand the interaction between PROTACs and the E3 ligase and/or target protein can aid in the rational design, screening, and optimization of drugs [14–16]. Virtual screening is an in silico technique aimed at identifying potential ligands of a biological target from a large database of chemical structures [17] Most virtual screening methods can be classified into ligand-based virtual screening or structure-based virtual screening. Ligand-based virtual screening analyzes the three-dimensional arrangement of key functional groups in the ligands to determine quantitative structure-activity relationships from a library of compounds [18, 19]. In contrast, structure-based virtual screening relies on knowledge of the 3D structure of a target of interest. Molecular docking has been one of the most commonly used types of structure-based drug design since the development of the first algorithms in the 1980s. Our group has discovered several protein–protein interaction inhibitors through in silico virtual screening, including CDK9–cyclin T1 protein–protein interaction inhibitors [20, 21], EED-EZH2 protein–protein interaction inhibitors [22], and Keap1–Nrf2 protein–protein interaction inhibitors [23].
Computational techniques have also been used to study the structures of protein-PROTAC-E3 ligase ternary complexes. In silico strategies can be used to analyze the possible interactions between target ligands and the E3 ligase protein, and between E3 ligase ligands and the target protein [24–26]. The simplest method for docking POI-PROTAC-E3 ternary complexes involves docking the PROTACs into the pocket of one protein, selecting a scalable docking pose, and then adding the second protein onto its corresponding binder [27, 28]. Several computational tools have been developed to model POI-PROTAC-E3 ternary complexes, including protein–protein docking [15], molecular dynamics [29], and direct analysis of the conformational structure of the PROTAC without the protein [30–32]. Pfizer has reported a computational workflow for assessing the steric compatibility between a PROTAC and two proteins [33]. In 2019, Williams et al. have reported an accurate computational modeling tool for POI-PROTAC-E3 ternary complexes that has been applied to multiple targets and E3 ligases as well as different PROTACs [26]. The ternary complexes were divided into two binary parts: the target protein and its ligand, and the E3 ligase-ligand complex. A PROTAC modeling tool based on the Molecular Operating Environment (MOE) was established, which involved docking these two parts and comparing the conformational ensemble of the user-supplied PROTACs with protein–protein docking. Furthermore, the same research group has provided two other PROTAC modeling methods to extend the design applications. One is a double-clustering approach that limits the PROTAC conformation at each binding moiety rather than the conformational ensemble, thereby requiring fewer PROTAC conformations and less computational time for modeling. Another method focuses on the shortest path between the target-ligase-binding moieties that are suitable for linker placement. User-supplied PROTACs could be added to the two binding moieties at the shortest path, to obtain higher hit rates with more accurate but lengthier simulations [34].
The PROTAC strategy has transitioned from academia to industry over the past 20 years, and numerous reviews have explored the theory and application of this protein degradation technology [35, 36]. However, to our knowledge, no review has focused on the application of molecular docking and virtual screening to discover new PROTAC drug lead compounds. In this review, we focus on the discovery and optimization of new PROTAC compounds via molecular docking and virtual screening techniques, particularly molecular docking. In addition, potential advantages, challenges, and perspectives in PROTAC discovery based on molecular docking and virtual screening are discussed.
2. PROTAC DRUGS BASED ON PEPTIDES DISCOVERED BY MOLECULAR DOCKING AND VIRTUAL SCREENING
The earliest PROTAC drugs used peptide moieties to engage E3 ligase. The first PROTAC bifunctional molecule was reported by Sakamoto et al. [1]. The angiogenesis inhibitor ovalicin, which covalently binds methionine aminopeptidase-2 (MetAP-2), is associated with an IκB-α phosphopeptide, which is recognized by the F-box, thus leading to recruitment of the E3 ubiquitin ligase β-TRCP [1]. The binding of the chimeric molecule to MetAP-2 results in its degradation via the UPS. Montrose and colleagues have also designed a peptide-based PROTAC targeting the hepatitis B virus X protein, a major hepatocellular carcinoma biomarker developed from hepatitis B virus [37]. The PROTAC was conjugated to a poly-arginine cell-penetrating peptide to improve cell permeability. This section discusses the application of molecular docking and virtual screening techniques to identify peptide-based PROTACs ( Table 1 ).
Structures and biological activities of peptide PROTACs based on molecular docking and virtual screening.
| Original name | Structure | Target protein | Disease | E3 ligase | Activity | Software | Ref. | |
|---|---|---|---|---|---|---|---|---|
| DC50 | Dmax% | |||||||
| Compound #8 |
 | Smad3 | Renal fibrosis | VHL | / | / | GLIDE molecular docking | [42] |
Mothers against decapentaplegic homolog 3 (Smad3) increases injury or inflammation in renal fibrosis, whereas knockout of Smad3 impedes fibrosis in animal models of kidney-injury nephropathy [38–40]. Hence, inducing Smad3 degradation may be a viable strategy to treat renal fibrosis. Under normal circumstances, the UPS only degrades phosphorylated Smad3 that is transported out of the nucleus [41]. Wang et al. have designed a new PROTAC through combining a hydroxylated pentapeptide of HIF1α, which acts as a specific recognition ligand of the E3 ligase VHL, with Smad3 ligands discovered through virtual screening from the Enamine compound library. They obtained 13 small-molecular compounds from the Enamine library via molecular docking with GLIDE, and found that the ligand EN300-72284 showed the best affinity, according to surface plasmon resonance analysis. The structure and degradation ability of the resulting PROTAC was confirmed by mass spectrometry and western blotting, thus demonstrating that this molecule might be useful to promote the degradation of Smad3 in preventing renal fibrosis [42].
However, peptide-based PROTACs can be limited by low activity and poor cell permeability, owing to their large molecular size [43]. Moreover, peptide-based PROTACs are sufficiently large to be recognized by the immune system and stimulate production of antibodies, thus decreasing their stability in humans. Consequently, small-molecule-based PROTACs have attracted attention for the development of clinical candidates.
3. PROTAC DRUGS BASED ON SMALL MOLECULES, DISCOVERED BY MOLECULAR DOCKING AND VIRTUAL SCREENING
Small-molecule-based PROTAC drugs use a small-molecule moiety for recognizing the E3 ubiquitin ligase. Small-molecule-based PROTACs provides several advantages over peptide-based PROTACs, including superior absorption, distribution, metabolism, and elimination properties [44]. The first small-molecule PROTAC was reported by Smith et al. for the degradation of AR. A polyethylene glycol–based linker was used to connect an AR ligand, called a selective AR modulator (SARM), with nutlin, a ligand of the MDM2 E3 ligase [45]. The synthesized SARM-nutlin PROTAC recruits AR to MDM2, thus resulting in the ubiquitination and degradation of AR by the proteasome. This section discusses the application of molecular docking and virtual screening to develop small-molecule-based PROTACs ( Table 2 ).
Structures and biological activities of small-molecule PROTACs based on virtual screening.
| Original name | Structure | Target protein | Disease | E3 ligase | Activity | Software | Ref. | |
|---|---|---|---|---|---|---|---|---|
| DC50 | Dmax% | |||||||
| (H-PGDS)-7 |
 | PGD2 | PGD2-related diseases | CRBN | 17.3 pM | 87.1 | MOE PROTAC-Modeling Tools | [3] |
| Compound 15 |
 | BRD4 and BRD2 | Acute myeloid leukemia | CRBN | / | / | PyMOL | [58] |
| MT-802 |
 | BTK | CLL | CRBN | 6.2 nM | 99 | / | [63] |
| SJF620 |
 | 7.9 nM | 95 | [64] | ||||
| YM181 |
 | EZH2 | Lymphomas | VHL | / | 80 | MOE 2014 | [71] |
| YM281 |
 | |||||||
| Compound SP4 |
 | SHP2 | Cervical cancer | CRBN | / | / | ICM-Pro 3.8.2 | [76] |
| Compound 16 |
 | EGFR | NSCLCs | VHL | 32.9 nM | 96 | MOE 2019.01 | [84] |
| Compound A16 |
 | AR | PCa | CRBN | / | 85 | MOE 2014 | [91] |
| Compound 6 |
 | 350 nM | / | GLIDE (Maestro 9.3 suite) | [92] | |||
| Compound 11c |
 | CDK9 | Breast cancer | CRBN | / | / | GOLD 5.1 | [97] |
Prostaglandin D2 (PGD2) is a major prostaglandin distributed mainly in the brain and mast cells in mammals [46]. The overproduction of PGD2 plays key roles in a variety of diseases, including allergic diseases [47], physiological sleep disorders [48], and Duchenne muscular dystrophy [49, 50]. PGD2 is produced by hematopoietic prostaglandin D synthase (H-PGDS); hence, H-PGDS may be a potential therapeutic target for PGD2-related diseases. Recently, researchers have successfully designed a chimeric PROTAC(H-PGDS)-1, which degrades H-PGDS via the UPS and effectively suppresses PGD2 production [51]. To optimize the degradation activity of PROTACs targeting H-PGDS, the authors performed a docking simulation of the ternary complex of PROTAC (H-PGDS), H-PGDS, and the E3 ligase cereblon (CRBN) with PROTAC Modeling Tools in MOE software ( Figure 3 ). The analogue PROTAC (H-PGDS)-7, which lacks a polyethylene glycol linker, has shown to have highly potent, selective, and effective H-PGDS degradation activity, and better in vivo activity, than that of the conventional H-PGDS inhibitor TFC-007 in a Duchenne muscular dystrophy model of mdx mice with cardiac hypertrophy ( Figure 3 ) [3].
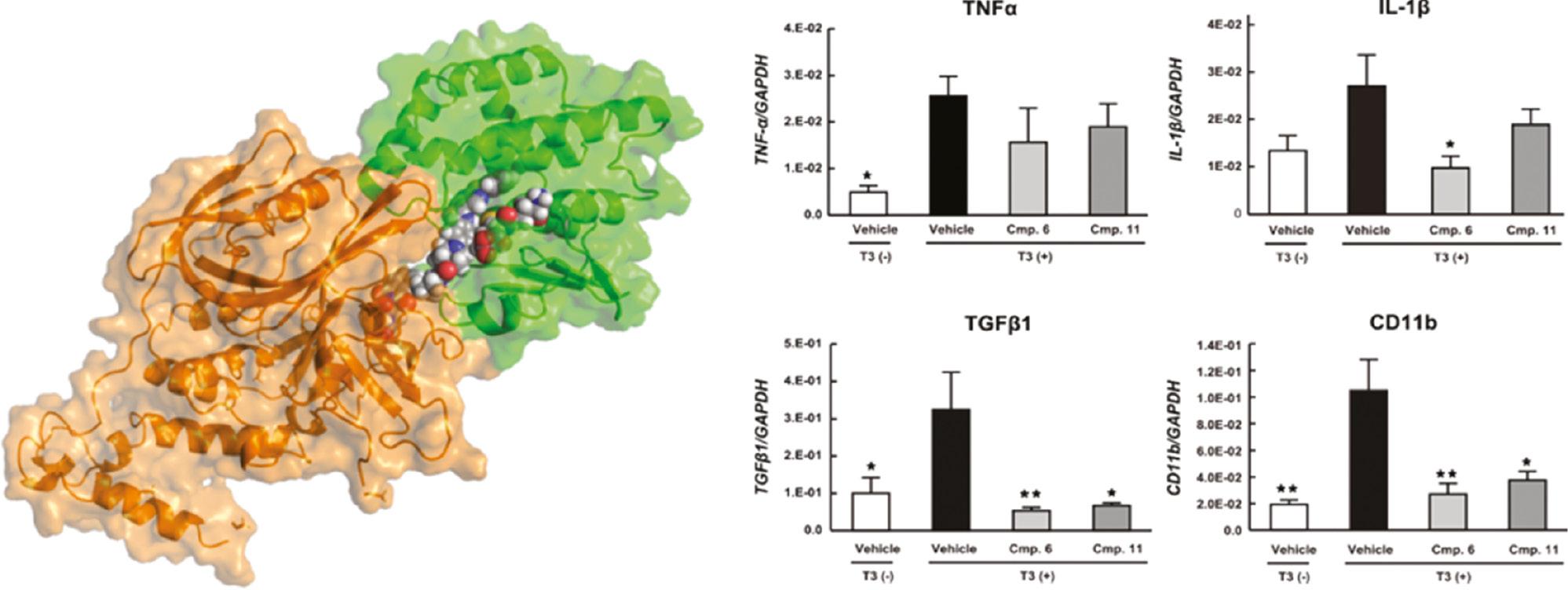
(Left) Docking research on the ternary complex of (H-PGDS)-7, H-PGDS (green), and CRBN (orange) with MOE software; (Right) PROTAC (H-PGDS)-7 decreases the mRNA levels of TNFα, IL-1β, TGFβ1, and CD11b in mdx mice with cardiac hypertrophy. The data in the bar graphs are the means ± SEM (n = 6–10). *P < 0.05 and **P < 0.01 compared with the T3-treated control in Dunnett’s test. Reproduced with permission from ref. [3]. Copyright 2021 American Chemical Society.
BRD2 and BRD4 belong to the bromodomain and extra-terminal domain (BET) family and are crucial targets for treatment of multiple diseases, owing to their effects on oncogenes [52], cytokines [53], and transcriptional regulation [54]. Pharmacologists have discovered several BET inhibitors according to the two bromodomains (BD1 and BD2) that exist in every BET protein structure. The BRD2 selective inhibitor ABBV-744 is already in clinical trials for the treatment of acute myeloid leukemia [55]. The first BET PROTACs were reported in 2015 [27], and degraders are increasingly being identified [56, 57]. However, the lack of intra-BET selectivity has limited their application for target verification and may also induce adverse effects or toxicity. A recent PROTAC BRD4 degrader, compound 15, has been reported to have 50-times-higher selectivity for BD1 than BD2 [58]. This compound was generated by attaching the CRBN/cullin 4A ligand thalidomide to a BET inhibitor with an 8-carbon chain linker. On the basis of docking analysis, the selectivity arises from the hydrogen-bonding interactions between the compound and Asn140, Asp144, and Leu92. This compound induces BRD4 degradation in leukemia cell lines. In the future, the bromodomains of BET proteins may serve as potential target domains for the virtual screening of intra-BET selective degraders.
Bruton’s tyrosine kinase (BTK), a Tec family kinase found in B-cells, promotes multiple pro-survival and proliferative pathways [59, 60]. Given its key role in promoting constitutive proximal B-cell receptor signaling in patients with chronic lymphocytic leukemia (CLL), BTK has been considered a potential drug target for CLL [61]. Drug resistance to ibrutinib—the most successful clinical BTK inhibitor, which irreversibly targets cysteine-481 in the ATP binding pocket of BTK – has been encountered, because mutation in cysteine-481 results in disease relapse [62]. Buhimschi and co-workers have developed a small-molecule PROTAC to inhibit both wild-type and C481S BTK. They used a reversible ibrutinib derivative to bind BTK and pomalidomide to engage the E3 ubiquitin ligase CRBN. Through docking of candidate structures with different linker regions to BTK, the researchers have successfully designed MT-802, the most potent BTK PROTAC, with an eight atom-linker ( Figure 4 ). MT-802 degrades the detectable BTK pool at nanomolar concentrations and exhibits fewer off-target kinase activities than ibrutinib ( Figure 4 ), even in cells isolated from patients with CLL [63]. Nevertheless, its poor clearance and half-life have restricted possibilities for its further development in vivo. Therefore, the researchers introduced structural modifications on the CRBN ligand of MT802, and designed a series of new PROTACs. On the basis of cell assays, compound SJF620 exhibits a better pharmacokinetic profile than MT802, and simultaneously retains potent degradation of BTK in mice [64].
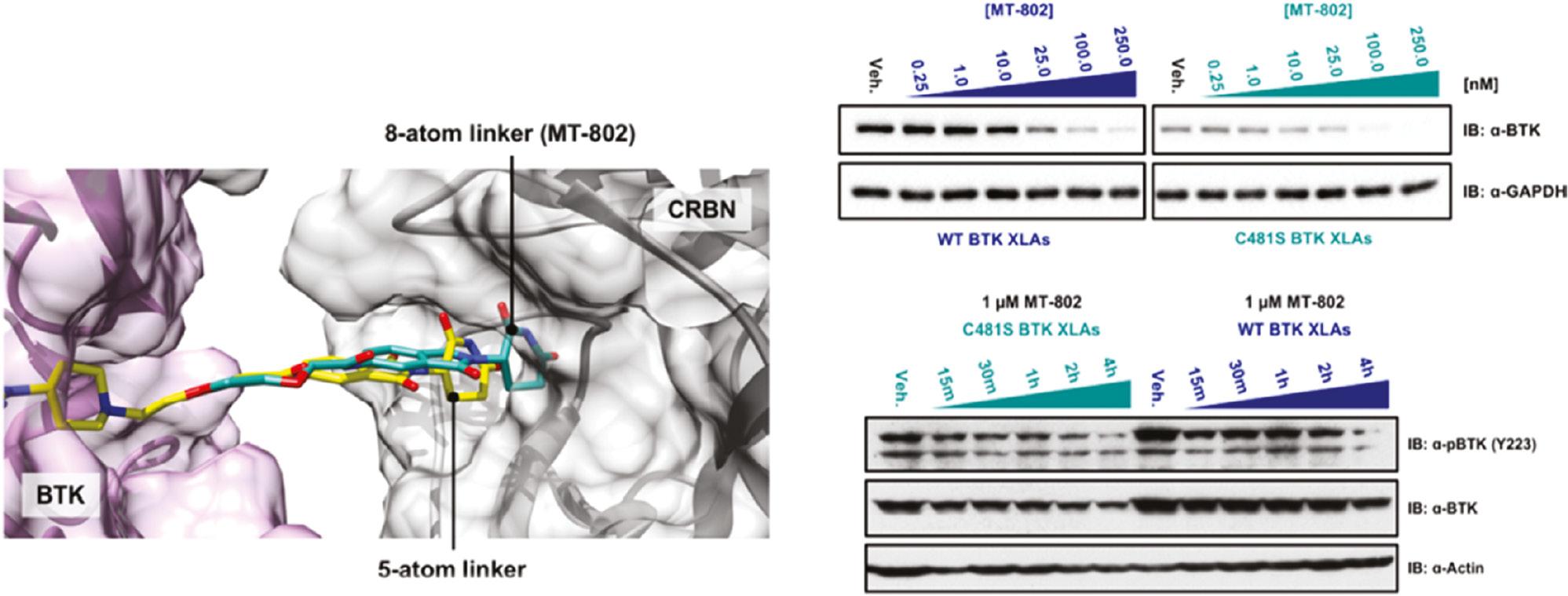
(Left) Docking study on MT-802 (green), BTK (5P9J, purple), and cereblon (gray); (Right) MT-802 degrades wild-type and C481S-mutant BTK. Reproduced with permission from ref. [63]. Copyright 2018 American Chemical Society.
EZH2 is the enzymatic subunit of Polycomb repressive complex 2 (PRC2), which mainly trimethylates lysine 27 of histone H3 and consequently silences gene transcription. EZH2 overexpression or gain-of-function mutations are associated with multiple cancers [65, 66]. Traditional EZH2 inhibitors focus on inhibition of methylation enzymatic activity; however, increasing evidence indicates that the protein itself is involved in tumor proliferation in a methylation-independent mechanism [67–69], thus causing acquired resistance and drug insensitivity [70]. Hence, the degradation of EZH2 protein might be a potential therapeutic method for EZH2-dependent cancers. Tu et al. have reported specific PROTAC degraders of EZH2, which have shown better antitumor effects against lymphomas in vitro and in vivo than traditional EZH2 inhibitors. The authors have selected the EZH2 inhibitor EPZ6438 as the target protein ligand, and designed and synthesized two series of PROTAC-based EZH2 degraders that recruit different E3 ligase systems, von Hippel–Lindau (VHL) or CRBN. Through in vitro experiments and molecular docking model analysis ( Figure 5 ) with EZH2 (PDB ID: 5LS6) in MOE, the authors successfully identified compounds YM181 and YM281 targeting VHL E3 ligase as the best EZH2 degraders [71].
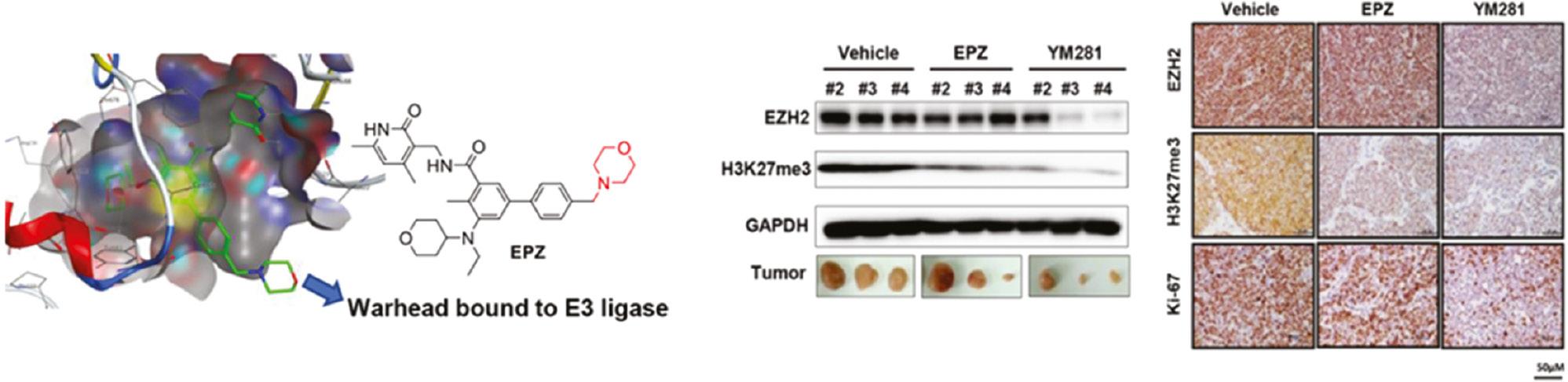
(Left) Docking conformation of EPZ6438 in the catalytic domain of EZH2 (PDB ID: 5 LS6) in MOE software; (Right) western blot analysis results and representative tumor images of EZH2 and H3K27me3 levels in representative SU-DHL-6 model excised tumors. Reproduced with permission from ref. [71]. Copyright 2021 American Chemical Society.
Mutations in Src homology region 2-containing phosphatase 2 (SHP2) are associated with multiple cancers [72]. SHP2 also participates in the regulation of the immune system, as a downstream effector of the PD-1 receptor [73, 74]. Although several SHP2 small-molecule inhibitors have been reported over the past two decades, the sequence similarity between SHP1 and SHP2 has hindered their further application. Targeting SHP2 with the PROTAC strategy might offer another avenue for impeding SHP2 activity in cancer treatment [75]. Zheng and co-workers have developed a PROTAC based on SHP099, a selective allosteric inhibitor of SHP2 activity. With a docking model of the SHP099-SHP2 complex, a series of new PROTACs have been designed by using the free amino group of SHP099 to target SHP2. The molecular docking results indicated that the designed PROTACs bind SHP2 through hydrophobic interactions. In in vitro assays, the PROTAC SP4 has shown 100-times-higher inhibitory activity than SHP099 against SHP2 in HeLa cells, through suppressing the SHP2-mediated RAS/MAPK signaling pathway [76].
Epidermal growth factor receptor (EGFR), a receptor for members of the EGF family, is a transmembrane tyrosine kinase protein involved in many human malignancies, particularly non-small-cell lung cancers (NSCLCs) [77–79]. EGFR tyrosine kinase inhibitors that inhibit the activity of mutant-EGFR ATP-binding domains have been approved by the FDA for the treatment of NSCLCs. However, drug resistance due to multipoint EGFR mutations has been observed [80]. EGFR-targeting PROTACs based on pomalidomide to degrade both EGFREx19del and EGFRL858R/T790M resistant proteins have been reported [81]. Aboelez et al. used molecular docking to design new pomalidomide-based EGFR-targeting PROTACs that can degrade both wild-type and mutant EGFR. The designed compounds have been docked against the ATP-binding sites of wild-type EGFR-TK (EGFRWT, PDB:4HJO) [82] and mutant EGFR-TK (EGFRT790M, PDB: 3W2O) with MOE ( Figure 6 ) [83]. On the basis of in vitro results ( Figure 6 ), compound 16 displays higher EGFR degradation efficacy than the other six compounds, through forming two hydrogen bonds with the Arg779 and Lys721 amino acids, thereby binding EGFRWT, and two hydrogen bonds with the Ser720 and Lys745 amino acids, thereby binding EGFRT790M; these findings support the key role of molecular docking in PROTAC screening [84].
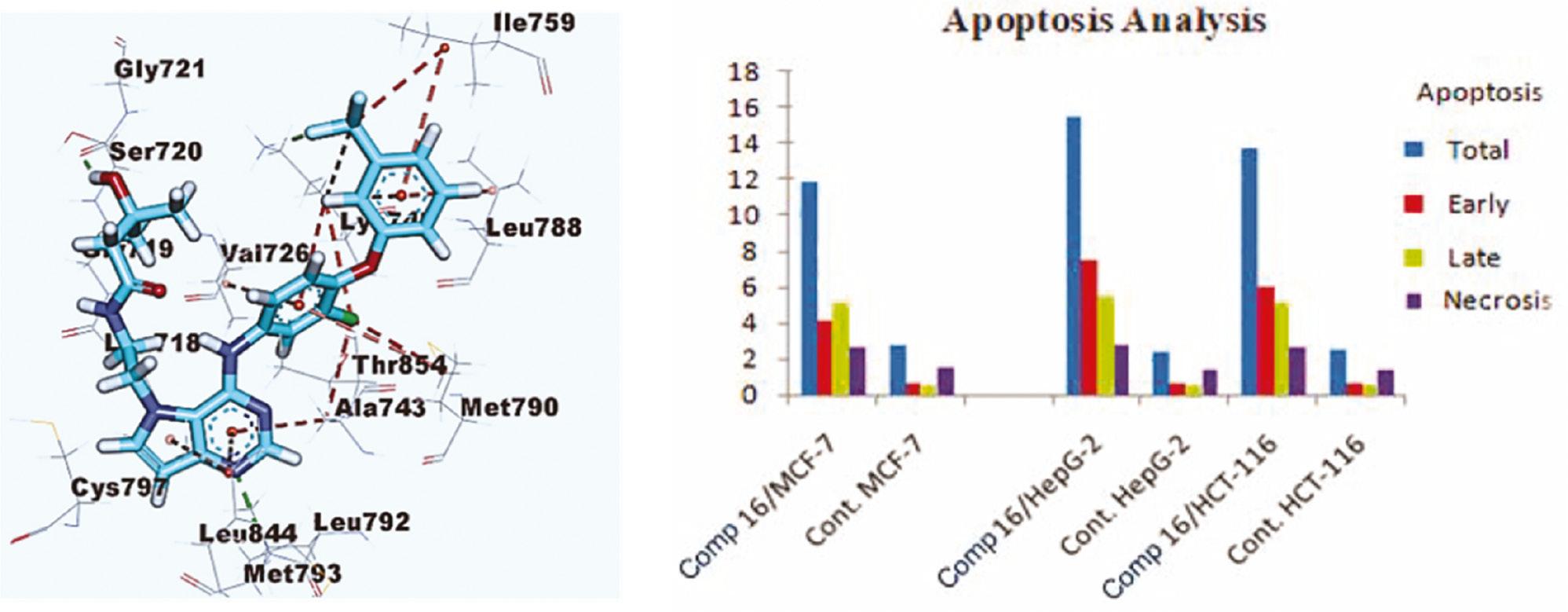
(Left) Docking model of TAK-285 with the active site of EGFRT790M in MOE software; (Right) apoptosis effects of compound 16 in different cell lines [84]. Copyright 2022 Journal of Enzyme Inhibition and Medicinal Chemistry.
Castration-resistant prostate cancer (CRPC) is resistant to androgen deprivation therapy, and thus is impervious to AR-antagonist treatment [85]. Recent research has revealed that AR overexpression is a biomarker for CRPC, particularly the AR-V7 splice variant, which contains an N-terminal and DNA-binding domain (DBD) but lacks ligand-binding domains [86–88]. Previous studies have indicated that small-molecule-based PROTAC with CRBN/cullin 4A neddylation degradation systems, such as the clinical phase 2 drug ARV-110 for prostate cancer (PCa), can degrade AR [89]. Liang et al. have designed and synthesized a group of phthalimide-based PROTAC compounds on the basis of the high-affinity AR agonist RU59063 [90]. Among all complexes, compound A16 showed the best AR binding affinity and AR degradation activity. To further verify the mechanism of AR degradation, the authors analyzed the docking model of compound A16 with AR protein (PDB: 2AXA) and the CRBN E3 ubiquitin ligase (PDB: 4CI3) in MOE, and found that A16 binds AR and E3 ubiquitin ligase through hydrogen bonding [91]. Bhumireddy et al. used proprietary computational algorithms and rational structure-activity-relationship optimization to select the best AR-V7 PROTAC degrader, compound 6, based on the AR DBD binder VPC-14228. This PROTAC degrader was designed by studying VPC-14228/AR-DBD/VHL ternary complex models in GLIDE (Maestro 9.3 suite) [92]. These docking models have provided guidance for designing a new PROTAC skeleton targeting AR for PCa.
Cyclin-dependent kinase 9 (CDK9) is a serine/threonine kinase involved in the expression of Mcl-1, an important survival protein for breast cancer cell growth [93, 94]. The inhibition or degradation of CDK9 has been found to effectively block the ability of cancer cells to resist apoptosis [95]. Several CDK9 inhibitors have been reported to treat advanced malignancies; and one of these inhibitors, flavopiridol, has entered clinical trials [96]. However, owing to the high sequence conservation of CDK9 with other CDKs, these inhibitors show reversible inhibition and drug resistance. Bian et al. have reported a CDK9 selective degrader (11c) with the CDK9 inhibitor wogonin [97]. They added substituent groups at position 8 of the wogonin flavone scaffold after preliminary structure-activity-relationship studies and molecular docking analysis between the CDK9 kinase domain and wogonin. Subsequently, wogonin and the CRBN ligand pomalidomide were conjugated via different linkers, and a series of wogonin-based PROTACs were synthesized. In western blotting assays, compound 11c showed the greatest ability to degrade CDK9. This compound also decreases the level of Mcl-1, and increases the death of CDK9-overexpressing MCF-7 cells. Molecular docking in GOLD 5.1 has been used to study the binding between the wogonin-based PROTACs and CDK9, and has revealed a similar binding mode to that of wogonin itself, thus validating the use of molecular docking in the design of mechanism-based PROTACs.
4. CONCLUSIONS AND PERSPECTIVES
Traditional drug discovery methods are characterized by their time-consuming nature (10−15 years), high cost (400–800 million US dollars), and low success rates [98–100]. The new PROTAC drug discovery platforms are no exception, and may be even more challenging to establish, because of the relatively high molecular weights (mostly >800 Da) of PROTAC compounds. In this context, rapid computer technology developments and the abundance of available structural, chemical, and biological data may enable the application of powerful virtual screening methods to lower the cost of drug discovery, including for PROTACs [101, 102]. Virtual screening methods, particularly molecular docking, are now routinely used in the drug discovery and design process to identify novel chemical scaffolds from large compound libraries, analyze drug-target binding, accelerate structure-activity-relationship analysis, and verify the prediction of adverse effects, among other applications [103].
In the past two decades, the astonishing progress and increased interest and investment in targeted protein degradation by both academia and industry have indicated that PROTAC technology is growing into a critical and effective therapeutic modality. The PROTAC platform avoids the problems of gene knockdown/knockout off-target effects of siRNA therapeutics, as well as the poor cell permeability of antibodies. More than 50 proteins have been targeted for degradation with PROTAC technology, including EZH2, ER, BTK, and BRD4 [58]. Two heterobifunctional PROTACs, ARV-110 and ARV-471 (with undisclosed structures), targeting AR and ER, respectively, have entered phase 1 and phase 2 clinical trials (NCT03888612 and NCT04072952) for prostate and breast cancer, respectively [104, 105].
This review summarized the recent applications of molecular docking and virtual screening for new PROTAC drug discovery. With the aid of molecular docking, PROTAC compounds have been identified against nine target proteins: Smad3, PGD2, BRD4, BTK, EZH2, SHP2, AR, CDK9, and EGFR. Molecular docking has played key roles in selecting the correct linker atom number, screening suitable target protein ligands, and characterizing the drug-protein-E3 ternary complex interactions.
In the future, several challenges must be overcome. Most studies described herein have validated PROTAC activity with in vitro experiments; however, in vivo data are required for further development of the compounds as clinical candidates for subsequent pharmaceutical research. Moreover, the degradation activity of PROTACs is dependent on the E3 ligase, which is unequally distributed across cell types and tissues [106]. Therefore, the distribution of the PROTAC in the body must be considered. Finally, diseases such as cancer may develop PROTAC resistance via different mechanisms, such as mutation [107]. Here, molecular docking may be able to help optimize candidates that are effective against both wild-type and mutant variants of a target, as has been demonstrated in some of the studies described in this review.
In our view, the roles of molecular docking and virtual screening for PROTAC drug discovery will only increase in the future with the continuing development of computing technologies and artificial intelligence [108]. Moreover, as target proteins as well as E3s are increasingly researched, the number of ligands that can be incorporated into the chimeric PROTAC platform will vastly expand. We believe that molecular docking and virtual screening, combined with the maturation of PROTAC development principles, will provide new directions for screening and identifying new PROTAC drugs to offer patients new treatment options beyond conventional therapeutics.