INTRODUCTION
Nucleoside 5′-triphosphates (NTPs) in which the ribose is specifically deuterated are valuable in structural and biochemical studies of nucleic acids. They can be used, for example, to reduce the complexity of NMR spectra [1] and to discern mechanistic details of nucleic acid cleavage [2].
Such studies provide information not otherwise obtainable. For example, we used specifically deuterated deoxynucleotides to probe the mechanism of DNA cleavage by the hydroxyl radical [3]. The ability to chemically synthesize specifically deuterated DNA allowed us to determine quantitatively which hydrogen atoms of deoxyribose are abstracted by the hydroxyl radical from duplex DNA. This work both elucidated the chemical and structural mechanism of a widely used chemical footprinting agent and contributed to a more detailed understanding of ionizing radiation-induced DNA damage.
Despite wide interest in the results of this work, the methods we described have seldom been used because of the difficulty of producing specifically deuterated NTPs by chemical synthesis [4]. Chemical routes to NTP synthesis require multiple steps that often are laborious and time-consuming. The consequence is that specifically deuterated nucleotides have been largely inaccessible to the structural and molecular biologists who would be most interested in using them.
A breakthrough in making specifically deuterated nucleotides more widely available came from Williamson and coworkers, who developed an enzymatic approach for the synthesis of deuterated ribonucleoside 5′-triphosphates from isotopically labeled glycerol or glucose [5–7]. Their scheme was able to produce milligram quantities of NTPs sufficient for preparing RNA by in vitro transcription for use in NMR spectroscopy. They showed that the enzymes did not have to be highly purified to be effective, which significantly reduced the effort and cost of NTP synthesis. Their synthesis is convenient, as NTPs are produced in a “one-pot” reaction that involves multiple enzymes. Williamson and coworkers used this approach to produce NTPs with multiply deuterated ribose residues, because they used commercially available uniformly deuterated glucose or glycerol in their synthetic protocols [6, 7].
While multiply deuterated NTPs are useful for NMR experiments, our earlier work [3] and the work of others [2] showed that selective deuteration at a specific deoxyribose carbon atom allows for the measurement of a deuterium kinetic isotope effect on hydrogen atom abstraction by a chemical probe of DNA structure. This experiment provides a chemical probe analysis of nucleic acid structure at single-atom resolution.
We describe here a comprehensive protocol, based on Williamson's pioneering work [6, 7], for the one-pot enzymatic synthesis and purification of milligram quantities of specifically deuterated adenosine 5′-triphosphate (ATP), guanosine 5′-triphosphate (GTP), uracil 5′-triphosphate (UTP), and cytidine 5′-triphosphate (CTP), for use in chemical probe experiments on RNA structure. Our protocol is designed to produce a series of NTPs each of which has deuterium incorporated at only one of the five carbon atoms of ribose, because our starting material is commercially available specifically deuterated ribose. We therefore are able to synthesize the complete set of 20 specifically deuterated ribonucleoside triphosphates, in which each ribonucleotide (ATP, UTP, CTP, or GTP) contains one of five deuterated riboses (either 1′-D, 2″-D, 3′-D, 4′-D, or 5′,5″-D2; see Figure 1A for ribose hydrogen numbering scheme). Reactions are complete in a few hours. Reaction progress is readily monitored by high-performance liquid chromatography (HPLC). NTPs synthesized using this protocol are greater than 98% isotopically pure at the site of deuterium substitution. A key advance that we detail in our protocol is the development of highly effective chromatographic purification of deuterated NTPs. We demonstrate that the purified deuterated NTPs are suitable for synthesis of RNA via in vitro transcription.
As a demonstration of the utility of specifically deuterated NTPs for RNA structural biology, we recently used this synthesis protocol to produce specifically deuterated NTPs for hydroxyl radical cleavage analysis of RNA structure [8]. By measuring the deuterium kinetic isotope effect on cleavage of RNA containing deuterated ribose, we quantified the reactivity of individual RNA hydrogen atoms, thereby increasing the resolution of the chemical probe experiment to the level of a single atom. We describe here in detail how to implement this experiment using specifically deuterated nucleotides.
Enzymatic synthesis of NTPs
To illustrate the synthetic scheme, in Figure 1 we show the synthesis of all four NTPs specifically deuterated at the 5′ carbon of ribose. Deuteration at other ribose carbons is achieved by substitution of the appropriate specifically labeled ribose in the first step of the synthesis.
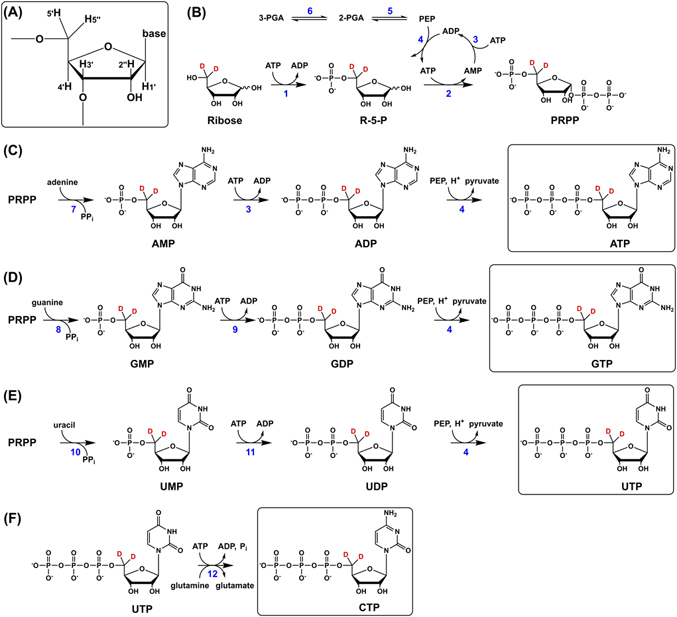
The synthetic routes for generating ATP, GTP, and UTP, which adhere closely to Williamson's schemes [6, 7], make use of a common intermediate, 5-phospho-d-ribosyl α-1-pyrophosphate (PRPP). In the first step, a specifically deuterated ribose (in Figure 1, d-[5′,5″-2H]ribose is used) is phosphorylated by ribokinase using ATP as cofactor to produce ribose-5-phosphate (R-5-P; Figure 1B). Next, R-5-P is pyrophosphorylated in a reaction catalyzed by PRPP synthetase to afford PRPP. The ATP consumed in the synthetase reaction is regenerated via an enzyme system consisting of pyruvate kinase (PK) and myokinase driven by phosphoenolpyruvate (PEP). PEP is generated from excess 3-phosphoglyceric acid (3-PGA) by the enzymes phosphoglycerate mutase and enolase.
The next step after the production of PRPP is the formation of a nucleoside monophosphate, which is achieved by adding the appropriate base (adenine, guanine, or uracil) and corresponding phosphoribosyltransferase enzyme (Figure 1C–E). For example, to synthesize adenosine monophosphate from PRPP, adenine and adenine phosphoribosyltransferase (APRT) are added to the reaction mixture (Figure 1C). The deuterated nucleoside monophosphate then undergoes consecutive phosphorylations, to yield first the nucleoside diphosphate and finally the desired triphosphate.
The synthesis of specifically deuterated cytidine 5′-triphosphate (CTP) is performed separately (Figure 1F) because CTP synthetase (CTPS) requires strict control of reaction conditions [7]. CTPS catalyzes the conversion of UTP to CTP using glutamine as a source of nitrogen and ATP as cofactor. GTP is included at low concentration since its presence stimulates the activity of CTPS.
Purification of deuterated NTPs
Because we found that the commonly used boronate affinity chromatography method is not effective under all circumstances encountered in this protocol, we developed two additional purification methods. Deuterated ATP, GTP, and UTP are purified using either boronate affinity or ion-exchange chromatography, and deuterated CTP is purified using ion-pair reversed-phase HPLC.
Boronate affinity chromatography makes use of the specificity of boronate toward cis-diol moieties, such as the C3′ and C2′ hydroxyls in ribose. Binding of a cis-diol to boronate is sensitive to temperature, so chromatography should be performed at 4°C. The resin is activated under alkaline conditions, and bound species are eluted using water acidified with CO2. The use of a flow adapter is not recommended since the resin bed volume collapses during the elution phase and gas is released, creating large bubbles that disturb the flow of mobile phase through the column.
While boronate resin is effective in binding ribonucleoside 5′-triphosphates, it is also capable of binding other components in the reaction mixture, such as Tris, glycerol, and unreacted ribose. This also becomes an issue when the reaction does not go to completion and substantial amounts of nucleoside 5′-mono- and diphosphates are still present.
Ion-exchange chromatography, using a volatile buffer salt such as ammonium bicarbonate, is effective in the purification of NTPs. This method has the advantage of being able to separate NTPs from the corresponding mono- and diphosphate intermediates. Ion-exchange chromatography is conveniently performed at room temperature with a flow adapter. An additional advantage is that diethylaminoethane (DEAE) resin typically costs significantly less than boronate affinity resin. A linear gradient of ammonium bicarbonate buffer (pH 9.5) is used to separate and elute the NTP from the resin. The final NTP product is obtained in the form of an ammonium salt. Since NTPs elute at higher salt concentrations than is typically observed in the boronate method, 2–3 rounds of lyophilization are required to remove residual salts.
A third purification method, which is primarily used for CTP, makes use of ion-pair reversed-phase HPLC on a semi-preparative scale. We found that the two methods of NTP purification described above are not suitable for CTP since they are unable to resolve the various triphosphate species required for CTP synthesis. Substantial quantities of ATP, GTP, unreacted UTP, and ADP are present in the final CTP synthesis reaction mixture. As we developed this method, a balance had to be struck between the high resolving power of HPLC and the constraint on column capacity inherent in using an analytical-scale instrument. While CTP is well resolved from interfering species, the capacity of the column we used is about 10 mg, so multiple injections were necessary to purify a typical reaction mixture. The method can be scaled up to accommodate the entire reaction mixture in a single injection provided that a preparative-scale HPLC instrument is available.
HPLC provides a real-time and unambiguous readout of which fractions contain the deuterated NTP of interest so that fractions can be pooled and concentrated immediately. The mobile phase additives triethylammonium acetate (TEAA) and acetonitrile are readily removed under vacuum. The final product is obtained as a triethylammonium salt. If residual TEA interferes with a downstream process, the purified NTP can be precipitated with ethanol and sodium acetate to afford the sodium salt.
Enzyme preparations
Although some of the enzymes used in the synthesis scheme (Figure 1) are commercially available, others must be expressed in E. coli and isolated. Partial purification is sufficient for many of the enzymes [6, 7]. Most of the enzyme preparations employ classical techniques such as ammonium sulfate precipitation and ion-exchange column chromatography. While effective, these techniques can be time-consuming, especially when multiple steps are required. All of the noncommercial enzymes have now been made available via expression vectors that code for enzymes containing a polyhistidine-tag (His-tag) [9, 10]. Because His-tagged enzymes can be purified directly from a cell lysate in a single, convenient, and efficient affinity chromatography step, their use will greatly streamline the synthesis protocol.
The enzymes that we used in this protocol are listed in Table 1.
| # | Name | Abbreviations | Native enzyme | His-tagged enzyme |
|---|---|---|---|---|
| 1 | Ribokinase | RK | [7, 11] | [9] |
| 2 | PRPP synthetase | PRPPS | [7, 11, 12] | [9] |
| 3 | Myokinase | MK | CA | |
| 4 | Pyruvate kinase | PK | CA | |
| 5 | Enolase | – | CA | |
| 6 | 3-Phosphoglycerate mutase (cofactor-independent) | iPGM | [13] | [10] |
| 7 | Adenine phosphoribosyltransferase | APRT | [7] | [9] |
| 8 | Xanthine–guanine phosphoribosyltransferase | XGPRT | [7] | [9] |
| 9 | Guanylate kinase | GK | CA | |
| 10 | Uracil phosphoribosyltransferase | UPRT | [6] | [9] |
| 11 | Nucleoside monophosphate kinase | NMPK | CA | |
| 12 | CTP synthetase | CTPS | [7, 14] | [9] |
Note: CA, enzyme is commercially available.
Enzymatic activity assays
The activities of purified iPGM, RK, APRT, and PRPPS are determined using variants of a continuous spectrophotometric assay that monitors the absorbance decrease at 340 nm as β-NADH is oxidized to β-NAD [9, 15].
For example, in the activity assay for iPGM (Figure 2), production of 2-phosphoglyceric acid (2-PGA) from 3-PGA is coupled to the oxidation of β-NADH via the actions of enolase, PK, and LDH [16]. Monitoring the decrease in absorbance at 340 nm over time yields the activity of iPGM, where one unit is defined as the amount of enzyme required to convert 1.0 μmol of 3-PGA per minute in the presence of 1.3 mM 2,3-diphosphoglycerate at pH 7.6 at 25°C.

The activity expressed in units per mL can be calculated using Equation (1):
where ΔA340 is the change in absorbance corresponding to the maximum linear rate of the reaction, Vtot is the total volume (in mL) of the reaction mixture, D is the dilution factor of the enzyme solution, Δt is the time (min) corresponding to the maximum linear rate of the reaction, 6.22 is the millimolar extinction coefficient of β-NADH at 340 nm, Venz is the volume (in mL) of enzyme solution added to the reaction mixture, and N is a stoichiometric coefficient equal to either 1 or 2 in our assays [11].Ribokinase activity is determined as described by Gross et al. [11] using the enzymes PK and LDH (Figure 3A) [7, 9]. In the purification of ribokinase, other enzymes that oxidize β-NADH may also be present and thus interfere with the assay. As a consequence, the final purified ribokinase solution is assayed with and without added d-ribose to determine the baseline rate of reaction.
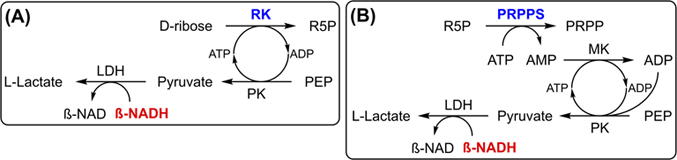
The activity of PRPPS is assayed using the protocol developed by Arthur et al. [9], in which the formation of PRPP from ATP and ribose-5-phosphate is coupled to the oxidation of β-NADH via the actions of myokinase, PK, and LDH (Figure 3B).
APRT activity is assayed [9] by using the enzyme to produce adenosine monophosphate (AMP) from PRPP and adenine (see Figure 1C). The AMP then enters the cycle shown at the bottom right of Figure 3B, ultimately resulting in β-NADH oxidation.
The activity of uracil phosphoribosyltransferase (UPRT) is confirmed by observing the formation of UMP from uracil and PRPP under conditions similar to those required for the synthesis of UTP (Figure 1E). After incubating the reaction mixture for a short time at room temperature, an aliquot is analyzed by HPLC for the presence of UMP. We found that the literature assay based on UV spectrophotometry [9] is not sufficiently sensitive for our purpose due to the large degree of spectral overlap between uracil and UMP.
To assay the activity of CTPS, conversion of UTP to CTP (Figure 1F) is monitored directly by HPLC at fixed time intervals. The reaction is set up on a 1-mL scale and uses standard (nondeuterated) UTP. For convenience, the reaction is set up in an HPLC vial which is placed in an autosampler at room temperature, and aliquots are injected directly into the HPLC system in an automated batch sequence.
To estimate the activity of xanthine–guanine phosphoribosyltransferase (XGPRT), the enzyme is used to synthesize GMP from PRPP in a small-scale version of the synthesis reaction that is shown in Figure 1D [7]. GMP production is monitored by HPLC.
Deuterated NTPs used in the synthesis of RNA via in vitro transcription: analysis of RNA structure by hydroxyl radical cleavage
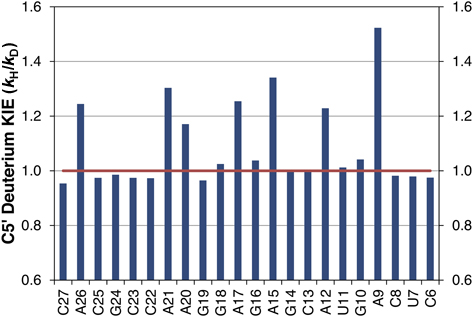
We demonstrate the suitability of deuterated ATP for the in vitro transcription of RNA by synthesis of the sarcin/ricin loop (SRL) RNA molecule. The SRL is a short (29 nt), biologically active RNA that is part of the 50S ribosomal RNA molecule [17–19]. The manageable size and numerous structural features of the SRL make it an attractive molecule to study using cleavage by the hydroxyl radical [8]. SRL RNA is synthesized using a commercially available in vitro T7 polymerase transcription kit in the presence of [5′,5″-2H]ATP to yield SRL RNA containing deuterium at the C5′ position of all adenine residues. The hydroxyl radical cleavage pattern of deuterated SRL RNA is compared to the cleavage pattern obtained from standard (nondeuterated) SRL RNA. The analysis reveals a substantial reduction in cleavage only at nucleotides that contain deuterium, indicating that RNA strand cleavage can be initiated by abstraction of a C5′ hydrogen atom from the ribose backbone.
Summary of deuterated NTP synthesis
Twenty specifically deuterated ribonucleoside 5′-triphosphate analogs were synthesized and used for hydroxyl radical cleavage studies of RNA. The complete set consisted of five deuterated analogs of each of the four NTPs (ATP, GTP, UTP, and CTP). The five deuterated analogs of ATP, for example, were of the form [X-2H]ATP, where X = 1′, 2″, 3′, 4′, or 5′,5″.
We found that synthesis of ATP, GTP, and UTP required substantially less 3-phosphoglyceric acid (3-PGA) than did the original protocols [6, 7]. In all cases, 3-PGA was initially added to each reaction mixture and another bolus was added 5 h after the reaction was initiated. NTP formation was complete after two additions of 3-PGA.
The volumes of reagents and enzymes used in the NTP synthesis reactions were scaled down relative to those in the original protocols [7]. In most cases, reactions were run on a 15-mL rather than a 50-mL scale, and no adverse effects on yield were observed. For each deuterated NTP synthesized, the yield was determined by HPLC analysis of the crude reaction mixture compared to an appropriate NTP standard. The theoretical yield was calculated from the amount of d-ribose starting material and the molecular weight of the NTP. For example, in the synthesis of [4′-2H]ATP, a total of 68 mg was expected based on an input of 22 mg d-[4′-2H]ribose. The yield determined for the synthesis of [4′-2H]ATP on a 15-mL scale was ∼65 mg, which corresponds to 96%. The yields determined for each set of NTP analogs, along with the total reaction times (in parentheses), were:
When boronate affinity chromatography was employed for purification, substantial amounts of the diphosphate product (and to a lesser extent the monophosphate product) co-eluted with the target NTP. For example, all GTP synthesis reactions contained 10–20% GDP relative to GTP. We found, though, that synthesis of RNA by in vitro transcription using purified deuterated GTP was successful despite the presence of large amounts of GDP, indicating a fair degree of tolerance of T7 polymerase for nucleoside 5′-monophosphate and 5′-diphosphate impurities present in NTP stock solutions.REAGENTS
Bacterial strains and plasmids for producing enzymes
Inducible E. coli strain for preparation of iPGM. We obtained this strain as a generous gift from Dr Mark J. Jedrzejas (Children's Hospital Oakland Research Institute, Oakland, CA).
Plasmid DNA constructs encoding for His-tagged UPRT and CTPS. We obtained these plasmids as generous gifts from Dr James Williamson (The Scripps Research Institute, La Jolla, CA).
Plasmid DNA construct encoding for His-tagged iPGM. We obtained this plasmid (pET21a) as a generous gift from Dr Clotilde Carlow (New England Biolabs, Ipswich MA).
Commercially available enzymes
Myokinase from rabbit muscle (MK; Sigma-Aldrich, cat. no. M3003)
Pyruvate kinase from rabbit muscle (PK; Sigma-Aldrich, cat. no. P7768)
Enolase from baker's yeast (Sigma-Aldrich, cat. no. E6126)
Guanylate kinase (GK; Sigma-Aldrich, cat. no. G9385)
Nucleoside monophosphate kinase (NMPK; Roche Applied Science, cat. no. 10107948001)
d-Lactic dehydrogenase (LDH; Sigma-Aldrich, cat. no. L3888)
Inorganic pyrophosphatase (New England Biolabs, cat. No. M2403S)
Lysozyme (Sigma-Aldrich, cat. no. L6876)
Antarctic phosphatase (AP; New England Biolabs, cat. no. M0289S)
T4 polynucleotide kinase (T4 PNK; New England Biolabs, cat. no. M0201S)
Reagents for enzyme preparation
One Shot BL21(DE3) Chemically Competent E. coli cells (Life Technologies, cat. no. C600003)
LB broth (Miller; Sigma-Aldrich, cat. no. L3022)
Ampicillin sodium salt (Sigma-Aldrich, cat. no. A0166)
3-Indoleacrylic acid (Sigma-Aldrich, cat. no. I2273)
2-Mercaptoethanol (BME; Sigma-Aldrich, cat. no. M3148)
Dithiothreitol (DTT; Sigma-Aldrich, cat. no. D0632)
Isopropyl β-d-1-thiogalactopyranoside (IPTG; Sigma-Aldrich, cat. no. I6758)
Streptomycin sulfate salt (Sigma-Aldrich, cat. no. S6501)
TALON Metal Affinity Resin (Clontech, cat. no. 635502)
Phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich, cat. no. P7626)
Precision Plus Protein Dual Color Standards (Bio-Rad, cat. no. 161-0374)
Reagents for enzyme activity assays, NTP synthesis, and NTP purification
Triethanolamine (TEA; Sigma-Aldrich, cat. no. T-1502)
Acetic acid (glacial; Sigma-Aldrich, cat. no. 320099)
5-Phospho-d-ribose 1-diphosphate pentasodium salt (PRPP; Sigma-Aldrich, cat. no. P8296)
d-(−)-3-Phosphoglyceric acid disodium salt (3-PGA; Sigma-Aldrich, cat. no. 79472)
Adenosine 5′-diphosphate sodium salt (ADP; Sigma-Aldrich, cat. no A2754)
Adenosine 5′-triphosphate disodium salt hydrate (ATP; Sigma-Aldrich, cat. no. A6419)
Guanosine 5′-triphosphate sodium salt hydrate (GTP; Sigma-Aldrich, cat. no. G8877)
l-Glutamine (Sigma-Aldrich, cat. no. G3126)
2,3-Diphospho-d-glyceric acid penta(cyclohexylammonium) salt (DPGA; Sigma-Aldrich, cat. no. D9134)
β-Nicotinamide adenine dinucleotide, reduced dipotassium salt (β-NADH; Sigma-Aldrich, cat. no. N4505)
Phospho(enol)pyruvic acid trisodium salt hydrate (PEP; Sigma-Aldrich, cat. no. P7002)
d-Ribose 5-phosphate disodium salt hydrate (R-5-P; Sigma-Aldrich, cat. no. R7750)
d-Ribose (Sigma-Aldrich, cat. no. R7500)
Adenine hydrochloride hydrate (Sigma-Aldrich, cat. no. A8751)
Guanine hydrochloride (Sigma-Aldrich, cat. no. 51031)
Uracil (Sigma-Aldrich, cat. no. U0750)
Cytosine (Sigma-Aldrich, cat. no. C3506)
Magnesium sulfate (MgSO4; Sigma-Aldrich, cat. no. M2643)
Manganese(II) chloride (MnCl2; Sigma-Aldrich, cat. no. 450995)
Potassium chloride (KCl; Sigma-Aldrich, cat. no. P9641)
d-[X-2H]ribose, where X = 1′, 2″, 3′, 4′, or 5′,5″ (Omicron Biochemicals, cat. nos. RIB-016, RIB-017, RIB-018, RIB-019, RIB-020)
Sodium phosphate monobasic (NaH2PO4; Sigma-Aldrich, cat. no. S3139)
Sodium phosphate dibasic (Na2HPO4; Sigma-Aldrich, cat. no. S3264)
Acetonitrile (HPLC-grade; Fisher Scientific, cat. no. A9981)
Affi-Gel boronate gel (Bio-Rad, cat. no. 153-6103)
Toyopearl DEAE-650M anion-exchange resin (Tosoh Bioscience, cat. no. 07473)
Reagents for RNA synthesis and analysis, and hydroxyl radical cleavage
MEGAShortscript T7 Transcription Kit (Life Technologies, cat. no. AM1354)
[γ-32P]ATP (3000 Ci/mmol, 10 mCi/ml; Perkin-Elmer, cat. no. BLU002A250UC)
Ammonium iron(II) sulfate hexahydrate ([(NH4)2Fe(SO4)2•6H2O]; Sigma-Aldrich, cat. no. 203505)
EDTA disodium solution (0.5 M; Life Technologies, cat. no. 15575-020)
(+)-Sodium l-ascorbate (Sigma-Aldrich, cat. no. A7631)
Hydrogen peroxide (H2O2, 30%; Sigma-Aldrich, cat. no. 95321)
Thiourea (Fisher, cat. no. T101-100) CAUTION: Potential carcinogen. May cause irritation to the eyes, skin and respiratory tract. Wear gloves, lab coat and safety goggles when handling thiourea solutions.
Milli-Q purified water or equivalent, autoclaved. Purified water is indicated in this protocol by dH2O. CRITICAL: The quality of water used in the preparation of reagents and buffers is very important. Organic contaminants in the water can interfere with the hydroxyl radical cleavage reaction, as can contaminating metal ions (such as iron or copper).
Sodium acetate, 3 M solution, pH 5.5 (Life Technologies, cat. no. AM9740)
Ethanol (100 and 70% vol/vol) chilled to –20°C
N,N′-tetramethylethylenediamine (TEMED; Sigma, cat. no. T9281) CAUTION: Wear gloves, lab coat, and safety goggles when handling TEMED solutions.
Ammonium persulfate (APS; Fisher, cat. no. BP 179)
Urea (Sigma-Aldrich, cat. no. U5378)
Sigmacote (Sigma-Aldrich, cat. no. SL-2)
Acrylamide/bis solution, 40%, 19:1 (Bio-Rad, cat no. 161-0144)
TBE buffer (10×; Life Technologies, cat. no. AM9863)
EQUIPMENT
Pipettors, 1–10, 2–20, 20–200, and 200–1,000 µl (models P10, P20, P200, and P1000, respectively; Gilson), and corresponding tips
Gel-loading tips, 0.4 mm (Rainin, cat. no. GT-250-4)
Microcentrifuge (e.g., Eppendorf model no. 5415 C)
Ultracentrifuge, rotor, canisters, and 0.5-L plastic bottles
HPLC system (e.g., Dionex, Ultimate 3000) equipped with an autosampler, 0.1–100 μL syringe, 4 solvent channels, temperature-controlled column compartment, UV/Visible multichannel wavelength detector, and an automated fraction collector (e.g., Dionex, AFC-3000)
Hypersil GOLD C18 preparative HPLC column (250 × 10 mm; Thermo Scientific, cat. no. 25005-259070)
DNAPac PA 200 analytical HPLC column (250 × 4 mm; Dionex Corporation, cat. no. 063000)
Amicon Ultra-15 centrifugal filter (10 kDa NMWL; Millipore, cat. no. UFC901024)
NucAway spin columns (Life Technologies, cat. no. AM10070)
Econo-Column glass chromatography column (2.5 × 20 cm; Bio-Rad, cat. no. 737-2522)
Flow adapter for Econo-Column (Bio-Rad, cat. no. 738-0017)
Gradient maker (C.B.S. Scientific, cat. no. GM-500)
Cuvette for UV-visible spectrophotometry
25-mL round bottom flask and stir bar
pH electrode
Medium or large inflatable balloon
Acrodisc syringe filter (0.2-μm pore size; Pall Corporation, cat. no. 4652)
Lyophilizer
Vertical gel electrophoresis apparatus for running SDS-polyacrylamide gels (e.g., Mini-PROTEAN Tetra Cell; Bio-Rad Laboratories, cat. no. 165-8002)
Vertical gel electrophoresis apparatus for running thin sequencing (denaturing) gels (e.g., model no. S3S T-Rex Aluminum Backed Sequencer; Owl Separation Systems)
Gel dryer
Side-arm flask and stopper, and access to laboratory vacuum or a water aspirator
Spacer set and 30-well comb (0.33 mm thick)
Power supply (5,000 V; e.g., PowerPac HV; Bio-Rad, cat. no. 164-5056)
Plastic wrap
Chromatography paper (Whatman, cat. no. 3030917)
Typhoon Trio Phosphorimager, including storage phosphor screens, exposure cassettes, and ImageQuant software (GE Healthcare, cat. no. 63-0035-62)
SAFA gel analysis software [20]. The software and documentation are freely available for download at http://safa.stanford.edu
PROCEDURES
Transformation of E. coli with the expression vector for an enzyme
Thaw one vial of One Shot BL21(DE3) competent E. coli on ice and then add 2 μl of a stock solution of the expression plasmid to the vial, mix gently, and let sit for about 20 min on ice. Heat-shock the bacteria for 30 s in a water bath set to 42°C, add 250 μl of Super Optimal broth with catabolite repression (SOC) medium (part of the kit), and incubate at 37°C for 1 h with shaking. Spread 5 and 20 μl of the transformed bacteria on separate LB agar plates (containing 100 μg/mL ampicillin if the plasmid encodes AmpR) and incubate overnight at 37°C.
Use individual colonies from the plates to inoculate two 5-mL starter cultures and incubate overnight at 37°C with shaking.
Transfer one overnight culture to 1 L of pre-warmed LB media containing 100 μg/mL ampicillin and incubate at 37°C with shaking.
To avoid the cell transformation step in the future, prepare a cell stock from the other culture by pelleting the cells in a centrifuge, resuspending the cell pellet in 0.5 mL of fresh LB media, mixing with an equal volume of glycerol, and storing at –80°C.
Preparation of His-tagged UPRT and CTPS
Incubate cells containing the appropriate expression vector for about 4 h with continuous shaking at 220 rpm until the absorbance at 600 nm is greater than 0.6 AU.
Induce expression of the enzyme by adding IPTG to a final concentration of 1 mM. Incubate an additional 4 h with shaking.
Harvest cells by centrifugation at 10,000g for 30 min at 4°C. Gently resuspend the pellet in 25 mL lysis buffer (50 mM Tris-HCl (pH 7.5), 250 mM NaCl).
Probe-tip sonicate cells, on ice, for three cycles of 10 s “on” followed by 10 s “off,” pause for 2 min, and complete 3 more cycles of 10 s “on” followed by 10 s “off.”
Pellet the cell debris by centrifugation at 30,000g for 30 min at 4°C. Collect the supernatant for chromatography.
Pack a chromatography column in the cold room with about 3 mL of His-tag affinity resin and equilibrate with lysis buffer containing 10-mM imidazole.
Apply the supernatant sample directly to the column and continue to wash with about five column volumes of lysis buffer containing 10-mM imidazole.
Concentrate the protein solution and exchange the buffer: transfer the purified protein solution to a centrifugal filter (10 kDa NMWL) and centrifuge at 4,000g in a swinging bucket rotor until about 1 mL remains in the upper chamber. Discard the flow-through, add 13 mL cold 100-mM potassium phosphate buffer (pH 7.4) to the upper chamber, and centrifuge at 4,000g until about 1 mL remains in the upper chamber. Repeat the dilution and centrifugation steps twice more.
Add an equal volume of glycerol, mix well, and store at –20°C for short times (<4 weeks) or –80°C for longer times.
UPRT enzyme activity assay
Prepare a 5-mL solution consisting of the reagents that are listed in Table 2.
Aliquot 1 mL of the reagent mixture into each of four fresh 1.5-mL tubes and let them sit for at least 5 min at room temperature.
Add 0, 1, 10, and 40 µL UPRT enzyme solution to each tube to initiate the reaction. Incubate the reaction mixture at room temperature for 1.5 h.
Perform an HPLC analysis (see “HPLC analysis of NTPs,” below) of each sample, including the blank, to confirm the production of UMP in each reaction.
CTPS enzyme activity assay
Directly in a 2-mL HPLC sample vial, prepare a 1-mL solution at room temperature consisting of the reagents that are listed in Table 3.
Add 10–40 µL CTPS enzyme solution to the vial. Incubate for 10 min at room temperature.
Meanwhile, set up an analytical HPLC batch run (see “HPLC analysis of NTPs,” below) that will perform periodic 10-µL injections over a 5-hr period.
Add 0.2 mL of 50-mM glutamine in water to initiate the reaction. Place the sample vial in the HPLC autosampler and start the batch run.
Use the peak areas of CTP and the injection time stamps to visualize the progression of the CTPS reaction (Figure 6).

Preparation of iPGM
Incubate cells that have been transformed with the expression vector for iPGM at 37°C for about 8 h with shaking until the A260 is >0.7 AU
Split the culture into two 0.5-L plastic bottles and chill on ice for 10 min. Centrifuge the cell suspension at 6,000g for 15 min at 4°C, discard the supernatant, and resuspend the cell pellet in 15-mL cold lysis buffer (50 mM HEPES (pH 7.4), 10% sucrose (wt/vol), 1 mM DTT). Transfer the sample to a fresh conical tube.
Add 6 mg lysozyme and 200 μL 0.1 M EDTA to the mixture and mix. Incubate the sample for 20 min at 37°C and then place on ice.
Probe-tip sonicate, on ice, for three cycles of 10 s “on” followed by 10 s “off,” pause for 2 min, and complete three more cycles of 10 s “on” followed by 10 s “off.”
Centrifuge the lysate at 32,000g for 1 h at 4°C and transfer the supernatant to a fresh conical tube.
Add MnCl2 to a final concentration of 1.2 mM and PMSF (stock solution in 2-propanol) to a final concentration of 100 μM.
Apply 1.6 mL 20% streptomycin sulfate solution (wt/vol) and mix well. Incubate the sample for 2 min at 45°C and then place on ice. Centrifuge the solution at 30,000g for 30 min at 4°C to remove the precipitate and transfer the supernatant to a fresh conical tube.
Under continuous and gentle stirring, slowly add enough solid ammonium sulfate to the sample solution to achieve 50% saturation. Continue stirring while allowing the sample to sit for an additional 10–20 min to allow for full equilibration. Centrifuge the sample at 20,000g for 20 min at 4°C and then transfer the supernatant to a fresh conical tube. Add enough ammonium sulfate to achieve 80% saturation, allow the sample to stand an additional 10 min, and then centrifuge again for 15 min. Resuspend the pellet in 5 mL cold Buffer A (50 mM HEPES (pH 7.4), 100 mM KCl, 1 mM DTT, 0.1 mM PMSF, 0.05 mM EDTA, and 0.25 mM MnCl2).
Transfer the sample solution to a dialysis sack with a 12,000 MW cutoff and dialyze against 2 L Buffer A at 4°C for 3 h. Replace the used buffer with 2 L fresh Buffer A and dialyze for additional 3 h.
Mix the sample solution with an equal volume of glycerol, mix well, and store at –80°C.
iPGM enzyme activity assay
In a 15-mL conical tube (wrapped in aluminum foil to protect from light exposure), prepare a 5 mL solution at room temperature containing the reagents that are listed in Table 4.
Add 66 U d-lactic dehydrogenase, 46 U PK, and 10 U enolase to the reagent mixture and equilibrate at room temperature for 5–10 min.
Meanwhile, prepare a 20-fold dilution of the iPGM enzyme solution in 80 mM triethanolamine buffer (pH 7.6), containing 100 mM KCl and 2.5 mM MgSO4.
Add 1.0 mL of the reaction mixture into a cuvette and blank the spectrophotometer at 340 nm.
Add 10 µL of the diluted iPGM solution to the cuvette to initiate the reaction. Record the absorbance at 340 nm every 5 s for a total of 5 min. Repeat the measurement twice with fresh reagent mixture and iPGM enzyme solution.
Determine the iPGM activity, in units per mL, of each replicate measurement using Equation 1 with N = 1. Report the final activity as an average of all replicates (Figure 7).

Preparation of PRPPS
Incubate cells that have been transformed with the expression vector for PRPPS at 37°C for about 7 h with shaking
Harvest the cells by centrifugation at 10,000g for 30 min at 4°C. Resuspend the pellet in 25-mL cold 50-mM potassium phosphate buffer (pH 7.5)
Probe-tip sonicate, on ice, for three cycles of 10 s “on” followed by 10 s “off,” pause for 2 min, and complete three more cycles of 10 s “on” followed by 10 s “off.”
Pellet the cell debris by centrifugation at 30,000g for 1 h at 4°C.
To the supernatant, add 2.5 mL of 20% streptomycin sulfate solution (wt/vol) and mix well. Incubate the sample for 2 min at 45°C and then place on ice. Centrifuge the solution at 30,000g for 30 min at 4°C to remove the precipitate. Transfer the supernatant to a fresh conical tube.
Under continuous and gentle stirring, slowly add enough solid ammonium sulfate to the sample solution to achieve 35% saturation. Continue stirring while allowing the sample to sit for an additional 10–20 min to allow for full equilibration. Centrifuge the sample, discard the supernatant, and resuspend the pellet in 25-mL cold 50-mM potassium phosphate buffer (pH 7.5).
Add 0.1 M acetic acid dropwise under continuous stirring at 4°C until the pH reaches 4.5. Centrifuge the sample and discard the supernatant. Resuspend the pellet in 20-mL cold 50-mM potassium phosphate buffer (pH 7.5).
Concentrate the sample and exchange the buffer using a centrifugal filter (10 kDa NMWL): centrifuge at 4,000g in a swinging bucket rotor until about 1 mL remains in the upper chamber, add 13 mL cold buffer, and centrifuge again until about 1 mL remains in the upper chamber. Repeat twice.
Collect the remaining liquid from the upper chamber and assay it for PRPPS activity.
PRPPS enzyme activity assay
In 100 mM triethanolamine buffer (pH 7.6), prepare a 10 mL stock solution of the reagents that are listed in Table 5.
Add 20 U each of the following enzymes: d-lactic dehydrogenase, PK, and myokinase.
Add a 20-µL aliquot of the PRPPS sample to initiate the reaction. Record the absorbance at 340 nm every 5 s for a total of 5 min.
Determine the PRPPS activity, in units per mL, of each replicate measurement using Equation 1 with N = 2. Report the final activity as an average of all replicates (Figure 8).

Preparation of RK
Incubate cells that have been transformed with the expression vector for RK for 4 h with continuous shaking at 220 rpm until the absorbance at 600 nm is greater than 0.6 AU.
Add 1 mL 3-indoleacrylic acid (25 mg/mL in ethanol) to induce the expression of ribokinase. Incubate the culture for an additional 8–10 h with shaking. After every 1–2 h of growth, remove a 1-mL aliquot and store at 4°C.
Perform SDS-PAGE analysis of the reserved samples to confirm the over-expression of ribokinase (32 kDa).
Harvest cells by centrifuging at 10,000g for 30 min at 4°C. Gently resuspend the cell pellet in 30-mL lysis buffer 2 (50 mM potassium phosphate buffer (pH 7.5), 2 mM BME).
Probe-tip sonicate, on ice, for three cycles of 10 s “on” followed by 10 s “off,” pause for 2 min, and complete three more cycles of 10 s “on” followed by 10 s “off.”
Pellet the cell debris by centrifugation at 30,000g for 30 min at 4°C. Transfer the supernatant to a fresh 50-mL conical tube.
Add 3.0 mL streptomycin sulfate solution (20%, wt/vol) and mix well. Incubate the sample for 2 min at 45°C and then place on ice. Centrifuge the solution at 30,000g for 30 min at 4°C to remove the precipitate. Transfer the supernatant to a fresh 50-mL conical tube.
Add ammonium sulfate to 80% saturation. Allow the sample to incubate at 4°C for at least 15 min. Centrifuge to pellet the precipitate. Resuspend the pellet in 10-mL lysis buffer 2.
Perform DEAE chromatography with a 500 mL linear gradient of 0–300 mM KCl in lysis buffer 2. Collect 5–8 mL fractions. Assay each column fraction for ribokinase activity (see “RK enzyme activity assay” below) and combine the appropriate fractions.
Concentrate the sample and exchange the buffer using a centrifugal filter (10 kDa NMWL): centrifuge at 4,000g in a swinging bucket rotor at 4°C until about 1 mL remains in the upper chamber, add 13 mL cold lysis buffer 2, and centrifuge again until 2–3 mL remains in the upper chamber. Add glycerol to 40% concentration.
RK enzyme activity assay
Prepare 1 mL of stock solution for each sample to be assayed, using the reagents that are listed in Table 6.
Add 10 U d-lactic dehydrogenase and 7 U PK per 1 mL stock solution. Incubate at room temperature for 5–10 min.
Add 1–20 µL of the prepared ribokinase sample to initiate the reaction. Record the absorbance at 340 nm every 5 s for 5 min.
Determine the ribokinase activity, in units per mL, of each replicate measurement using equation 1 with N = 1. Report the final activity as an average of all replicates.
Preparation of APRT and XGPRT
We prepared APRT and XGPRT using the methods detailed by Tolbert and Williamson [7]. Briefly, an overproducing strain was induced with IPTG, cells were harvested, and ammonium sulfate precipitation and DEAE column chromatography were used to isolate the enzyme.
APRT enzyme activity assay
In a 15-mL conical tube (wrapped in aluminum foil to protect from light exposure), prepare a 5-mL solution at room temperature containing the reagents that are listed in Table 7.
Add 10 U d-lactic dehydrogenase, 10 U PK, and 10 U myokinase to the 5-mL reagent mixture and equilibrate at room temperature for 5–10 min.
Meanwhile, prepare an APRT enzyme solution in 50 mM Tris-HCl buffer (pH 7.8), containing 10 mM MgCl2.
Add 1.0 mL of the reaction mixture into a cuvette and blank the spectrophotometer at 340 nm.
Add 10–20 µL of the prepared APRT solution to the cuvette to initiate the reaction. Record the absorbance at 340 nm every 5 s for a total of 5 min. Repeat the measurement twice with fresh reagent mixture and enzyme solution.
Determine the APRT activity, in units per mL, for each replicate measurement using Equation 1 with N = 2. Report the final activity as an average of all replicates (Figure 9).

XGPRT enzyme activity assay
In a 15 mL conical tube, mix the reagents that are listed in Table 8 to give a final volume of 2 mL.
Add 1 U inorganic pyrophosphatase and 1 mg guanine.
Initiate the reaction by adding 20–30 μL XGPRT.
Over a period of 10 min, remove 100-μL aliquots from the reaction mixture for every 2 min.
Filter each aliquot and transfer to an HPLC sample vial.
Chromatograph the samples using reverse-phase HPLC (Figure 10).
Quantify the peak corresponding to GMP to estimate the activity of the enzyme.

ATP, GTP, and UTP synthesis
Weigh the reagents listed in Table 9 into a clean, dry 25-mL round bottom flask.
Add 14.5 mL potassium phosphate buffer and a stir bar to the flask and begin stirring gently.
Add 0.3 mL 0.5 M MgCl2 to the flask.
Adjust the pH dropwise to 7.6 with 1 M KOH.
Remove oxygen gas from the solution in the fume hood by bubbling with nitrogen gas for 15–20 min. TIMING: Work quickly to minimize the reintroduction of oxygen into the reaction mixture.
Add the enzymes listed in Table 10 to the reaction mixture.
Check the pH and adjust to 7.6 if necessary. Transfer a 50-μL aliquot of the reaction mixture to a fresh 1.5-mL microcentrifuge tube and store at –20 or –80°C. This sample will be used later for HPLC analysis.
Initiate the reaction by adding 10 μL of a 75 mM ATP stock solution, or, if applicable, the same deuterated analog of ATP that is being synthesized. Remove a 50-μL aliquot and store it in a –20°C freezer for later HPLC analysis. Inflate a balloon with nitrogen gas and place it over the neck of the round bottom flask. Create a seal by covering with a few layers of paraffin film.
After 1 h, check the pH and adjust to 7.6, if necessary. Reserve a 50-μL aliquot for HPLC analysis.
After 2 h, add 170 mg 3-PGA disodium and adjust the pH to 7.6, if necessary. Reserve a 50-μL aliquot for HPLC analysis. For GTP synthesis, add 2 U guanylate kinase. For UTP synthesis, add 1 U NMPK. NOTE: Check the pH of the reaction mixture periodically and adjust to 7.6 if necessary. Reserve an aliquot of the reaction mixture and store in the freezer at various time points (e.g., 1, 2, 5, 10, and 24 h) as needed for HPLC analysis.
After 5 h, add 170 mg 3-PGA to the reaction mixture and continue stirring.
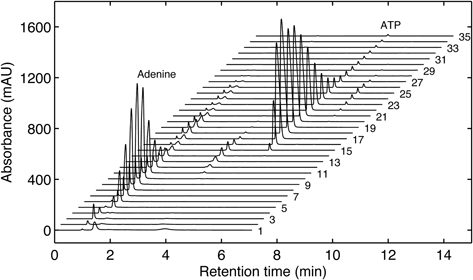
Continue stirring until the reaction has gone to completion, which can typically take between 5 and 24 h, as determined by HPLC (Figure 11).
Remove proteins and particulates: transfer the crude reaction mixture to a centrifugal filter (10 kDa NMWL) and centrifuge at 4,000g in a swinging bucket rotor for 15 min or until all liquid has filtered through to the bottom of the tube. CRITICAL: Make sure the centrifuge is properly balanced during operation.
Prepare an ethanol/dry ice bath in a heavily insulated vessel (such as a Dewar or Styrofoam box). Immerse the sample tube into the ethanol/dry ice bath at an angle. Rotate until all of the sample liquid is frozen solid. Uncap the tube, cover with two layers of paraffin film and poke several small holes in the film using a fine-gauge needle. Lyophilize the sample overnight or until the sample is dry. Store at 4°C for short times (<1 day) or –20°C or below for longer times.
| Reagent | Amount (mg) |
|---|---|
| d-[X-2H]ribose | 20 |
| DTT | 23 |
| 3-PGA | 170 |
Note: X = either 1′, 2″, 3′, 4′, or 5′,5″.

CTP synthesis
In a 20-mL glass scintillation vial, add the reagents listed in Table 11 to 5 mL of 50 mM Tris-HCl buffer (pH 7.8).
Add a stir bar to the vial and begin stirring. Remove a 50-μL aliquot for HPLC analysis and store at –20°C.
Initiate the reaction by adding glutamine to a final concentration of 1 mM.
Remove a 50-μL aliquot each hour. Store at –20°C for later HPLC analysis.
After 10 h, or when the reaction is complete as determined via HPLC analysis, syringe-filter the solution into a fresh scintillation vial through a filter with pore size 0.45 μm. Flash freeze the filtered solution in an ethanol/dry ice bath. Lyophilize overnight or until all water has been removed.
HPLC analysis of NTPs
In 200-µL conical HPLC sample vials, prepare 0.5 mM solutions of standard and deuterated ribonucleoside 5′-triphosphates in Milli-Q-purified water (≥18.2 MΩcm).
Prepare 1 L each of the following HPLC-grade mobile phases: water, 1.0 M NaCl, and 0.1 M NaOH. Filter the NaCl and NaOH solutions through a 0.45 μM membrane. Set up the solvent channels as (A) water, (B) 1.0 M NaCl, and (C) 0.1 M NaOH.
Equilibrate the HPLC system (DNAPac PA 200 analytical column) at a flow rate of 1.4 mL/min with 92.5% A, 5% B, and 2.5% C, at a column temperature of 30°C. Monitor the baseline absorbance at 260 nm.
Set up a batch run to perform 1–40 µL injections of a blank water solution, three consecutive injections of an NTP standard solution, at least two injections of a deuterated NTP solution, and one final standard NTP injection.
For each sample, pre-equilibrate the column for 5 min, inject the sample, run a linear gradient from 5% to 40% B (with constant 2.5% C) from 0 to 10 min, increase to 80% B, and hold for 3 min.
PURIFICATION OF DEUTERATED NTPs
Boronate affinity column chromatography
Set up a glass chromatography column in the cold room (a flow adapter is not recommended). Suspend 3 g Affi-Gel boronate affinity resin in 50-mL cold 0.5 M ammonium bicarbonate buffer (pH 9.5), let sit for 10 min, and then pack the resin into the column while maintaining a steady flow. Collect the flow-through into a designated waste container. Equilibrate the column with at least five column volumes of cold 0.5 M ammonium bicarbonate buffer (pH 9.5). CRITICAL: Perform boronate affinity chromatography at 4°C, as resin binding affinity can be drastically weakened at higher temperatures.
Meanwhile, dissolve the lyophilized NTP residue in 10 mL of cold 0.5 M ammonium bicarbonate, let it sit for 5–10 min, and then filter the solution using a syringe filter (0.45 μm) into a fresh conical tube. NOTE: Filtering samples will prolong the lifetime of the affinity resin and column.
Apply the sample directly to the top of the affinity resin. Continue washing the column with cold 0.5 M ammonium bicarbonate. Monitor and record the absorbance value at 260 nm.
When the A260 drops below ∼0.1 AU, elute the bound material with cold acidified dH2O. Collect 5 mL fractions. Combine fractions having an absorbance >0.1 AU and transfer to a clean, dry lyophilization flask. Flash freeze the sample in an ethanol/dry ice bath. Lyophilize overnight or until all water has been removed.
Resuspend the residue in a minimal amount of dH2O (∼5 mL), making sure to rinse the flask walls to maximize recovery of solute. Repeat the flash freezing and lyophilization process.
Ion-exchange column chromatography
Set up a glass chromatography column on the benchtop at room temperature. Pour, in one motion, ∼30 mL DEAE-650M resin into the column with the stopcock in the open position. Continue to add 5 mM ammonium bicarbonate (pH 9.5) while collecting the flow-through in a large marked waste container. Equilibrate the column with at least five column volumes of 5 mM ammonium bicarbonate. NOTE: The use of a flow adapter is recommended.
Meanwhile, prepare the NTP sample by resuspending the lyophilized residue in 15 mL of 50 mM ammonium bicarbonate, let sit for 5–10 min, and pass through a syringe filter with a pore size of 0.45 μm. NOTE: Filtering samples before loading them onto the column will prolong the lifetime of the column and resin.
Load the sample into the flow adapter, wash the column with one column volume of 5 mM ammonium bicarbonate, and begin a linear gradient of ammonium bicarbonate buffer (pH 9.5) from 5 to 500 mM. Collect 5 mL fractions. Determine via HPLC which fractions contain the deuterated NTP of interest (Figure 12).
Combine the appropriate fractions in a clean, dry lyophilization vessel, flash freeze in an ethanol/dry ice bath, and lyophilize overnight or until all liquid has been removed.
Resuspend the residue in a minimal amount of dH2O and repeat flash freezing and lyophilization. TIP: A third round of resuspension and lyophilization is sometimes necessary if there is too much residual salt.
Ion-pair reversed-phase HPLC
Turn on the UV detector, set the column temperature to 30°C, and equilibrate the reverse-phase column with a mobile phase of 25 mM TEAA buffer (pH 6.5) at a flow rate of 5.0 mL per min. Set up a linear gradient from 0 to 8% acetonitrile in 25 mM TEAA buffer (pH 6.5) over 16 min.
Meanwhile, dissolve the lyophilized residue in 2.0 mL or less of mobile phase, let the sample sit for 5–10 min, and then pass the solution through a syringe filter with a 0.45-μm pore size.
Manually inject 250 μL of a mobile phase blank solution and observe the baseline absorbance at 260 nm.
Perform at least three manual injections of 250 μL of an NTP standard solution.
Perform as many 250-μL sample injections as needed until the sample is consumed. Collect every fraction that contains the NTP of interest (see Figure 13).
Pool the appropriate fractions into a lyophilization flask, flash freeze in an ethanol/dry ice bath, and lyophilize overnight or until the sample is dry.

MASS SPECTROMETRY OF PURIFIED NTPs
In 200 µL HPLC conical sample vials, prepare 0.1 mg/mL solutions of standard and purified deuterated ribonucleoside 5′-triphosphates in high quality water (≥18.2 MΩcm). Prepare a blank sample containing the same water used to dissolve the NTPs.
Set up an HPLC-MS instrument to perform 10 µL sample injections directly into the mass spectrometer operating in positive-ion mode. Use a mobile phase of 0.1% formic acid in water with a flow rate of 0.2 mL per min.
Inject 10 µL of the water blank before and after each NTP sample injection to eliminate crossover contamination.
Verify the accuracy of the spectrometer using the mass spectrum of the standard NTP solution. Compare the experimental mass-to-charge ratio to that expected for the standard and deuterated NTP (Figures 14 and 15).


USE OF DEUTERATED NUCLEOTIDES AS PROBES OF RNA STRUCTURE
Preparation of RNA by in vitro transcription
Prepare a stock solution of a deuterated NTP (75 mM) in RNase-free water.
Set up two 20-µL-scale transcription reactions according to the T7 MEGAShortscript kit instructions. For one of the samples, substitute 2 µL deuterated NTP solution in place of the appropriate stock solution included with the kit.
Incubate for at least 3 h at 37°C. Add 1 µL TURBO DNase and incubate for an additional 15 min at 37°C.
Filter each reaction mixture through a NucAway spin column according to the manufacturer's instructions. Elute in 40-µL RNase-free water.
Add 4-µL ammonium acetate solution (5 M) to each tube and mix. Precipitate the RNA by adding 150 µL cold 100% ethanol and mix well. Incubate the samples at –80°C for 1 h.
Centrifuge the precipitated RNA samples at 20,000g for 30 min at 4°C.
Using a 200-µL pipet tip, carefully withdraw the supernatant without disturbing the pellet, which contains the RNA.
Wash the pellet with 200 µL of cold 70% ethanol. Centrifuge at 13,000g for 15 min at 4°C.
Discard the supernatant. Dry the pellets either on the benchtop or under vacuum.
Resuspend the RNA pellets in 10 µL RNase-free water.
5′-end labeling of RNA with 32P
Phosphatase reaction, to remove the 5′-phosphate: In a 1.5-mL microcentrifuge tube, prepare 19 µL of a solution containing 50 pmol RNA in 1× AP buffer. Add 1 µL AP enzyme to the reaction and incubate for 30 min at 37°C. Heat the sample for 5 min at 65°C to inactivate the enzyme.
T4 PNK reaction, to add 5′-32P: To the reaction tube containing dephosphorylated RNA, add 11 µL RNase-free water, 5 µL T4 PNK buffer (10×), 1 µL T4 PNK enzyme, and 2.5 µL [γ-32P]ATP to give a final volume of 40 µL. Incubate the sample for 1 h at 37°C.
Add 40 µL Gel Loading Buffer II (from the T7 MEGAShortscript transcription kit) and mix well. Purify radiolabeled RNA by electrophoresis on a 12–15% denaturing PAGE gel.
Excise the gel band containing radiolabeled RNA and place into a fresh 1.5 mL microcentrifuge tube. Using a pipet tip, crush the gel slice against the wall of the tube. Elute the RNA by soaking in 150 µL buffer (25 mM phosphate (pH 7.6), 0.3 M sodium acetate, 0.1 M KCl) for 1 h at 4°C.
To remove polyacrylamide gel pieces, transfer the RNA solution to a 1.5 mL centrifugal filter (Ambion) and centrifuge for 5 min at 20,000g.
Ethanol precipitate and dry the RNA sample.
Hydroxyl radical cleavage reaction
In separate 1.5-mL microcentrifuge tubes, place 40 kcpm of singly end-labeled nondeuterated (ND) and deuterated RNA in a total volume of 7 µL of potassium phosphate buffer (pH 7.6). Also prepare one ND and one deuterated control RNA sample.
Renature the RNA by incubating the samples at 90°C for 2 min and then cooling on the benchtop for 10 min.
To initiate the hydroxyl radical cleavage reaction for the experimental samples, carefully spot 1 µL each of the following three reagent solutions as separate drops on the wall of the sample tube: 10 mM sodium ascorbate, 1.2% H2O2, and iron(II)-EDTA (1 mM iron(II)/2 mM EDTA). For the control samples, substitute RNase-free water for each of the three hydroxyl radical reagents. CRITICAL: Hydroxyl radical cleavage reagents should be freshly prepared. Reagents used to carry out hydroxyl radical cleavage should not be premixed before being added to the RNA sample. Premixing the reagents initiates the Fenton reaction prematurely, expending hydroxyl radicals before the target RNA is added.
With a flick of the wrist or a quick spin in a benchtop centrifuge, quickly add the cleavage reagent drops from the tube wall to the sample and mix gently. Immediately begin timing the reaction. Let the reaction proceed for 2 min, and then quench by adding 1 µL of 100 mM thiourea. NOTE: The reaction time can be varied (1–5 min) to optimize the extent of RNA cleavage. Separate reactions should be carried out in parallel to determine the reaction time that gives optimum cleavage. CRITICAL: The Fenton reaction is initiated by mixing the hydroxyl radical reagents, which generates the radical species necessary for cleavage of the RNA backbone. It is important to stop the cleavage reaction by adding the thiourea stop solution, which quenches the reaction by reacting with any remaining hydroxyl radicals.
Add 4 µL ammonium acetate solution (5 M) to each tube and mix. Precipitate the RNA by adding 150 µL cold 100% ethanol and mix well. Incubate the samples at –80°C for 1 h.
Centrifuge the precipitated RNA samples at 20,000 g for 30 min at 4°C.
Using a 200-µL pipet tip, carefully remove the supernatant without disturbing the pellet, which contains the RNA. Check the tube using a Geiger counter to ensure that a substantial fraction (>60%) of the radioactivity is retained in the pellet after discarding the supernatant.
Wash the pellet with 200 µL of cold 70% ethanol. Centrifuge at 13,000g for 15 min at 4°C.
Discard the supernatant and dry the pellets either on the benchtop or under vacuum.
Resuspend the RNA pellets in 4-µL Gel Loading Buffer II (from the T7 MEGAShortscript transcription kit) and analyze using high-resolution denaturing PAGE.
Denaturing polyacrylamide gel electrophoresis
Siliconize the larger of the two glass plates with 0.5-mL Sigmacote.
Assemble the gel plate sandwich with spacers and comb (0.33-mm thickness).
Prepare 70 mL of a 20% acrylamide:bisacrylamide (19:1) solution containing 7–8 M urea. Degas the solution for 5 min under vacuum. A 20% gel is appropriate for short RNAs, like the SRL. Use a lower gel percentage if studying a larger RNA molecule.
Initiate gel polymerization by adding 250 µL 10% (wt/vol) APS and 40 µL TEMED to the acrylamide/urea solution. Pour the gel and allow to polymerize for ∼ 1 h.
Mount the gel in the gel electrophoresis apparatus. Pre-run the gel at constant power (65 W) for at least 30 min. CRITICAL: The power at which the gel runs should be adjusted so that the surface temperature of the gel plates does not exceed 55°C.
Heat the RNA samples at 95°C for 2 min to denature the RNA. Load all 4 µL of each sample into a lane of the pre-run gel.
Electrophorese at constant power (65 W) for sufficient time to resolve the region of interest (∼3.5 h). The bromophenol blue and xylene cyanol dyes that are included in the gel-loading buffer provide convenient visible markers for estimating the optimum time for electrophoresis.
After electrophoresis, allow the gel to cool to room temperature and remove from the apparatus.
Place the gel sandwich on a lab bench and carefully remove one glass plate (the plate that was siliconized) from the gel assembly. Place a layer of plastic wrap on the exposed gel while trying to minimize wrinkles in the plastic.
Invert the assembly. Pull the plastic away from the glass plate slowly and carefully, peeling the gel along with the plastic.
Cover the exposed side of the gel with Whatman chromatography paper. Take care to avoid wrinkles between the gel and the Saran wrap. The presence of wrinkles can lead to inefficient gel imaging during exposure to the phosphor screen.
Dry the gel using the gel dryer for about 2 h at 80°C. The gel drying conditions should be optimized for the acrylamide percentage of the gel and the gel dryer that is used. The drying conditions may be varied by adjusting the drying time or the length of time that the vacuum is applied to the gel.
Place the dried gel in an exposure cassette at room temperature and expose to a phosphor screen. The exposure time can vary from less than 1 h to 18 h depending upon the level of radioactivity of the RNA sample.
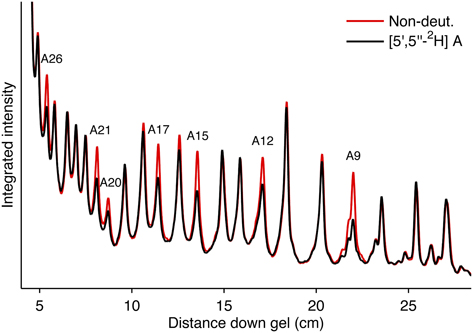
Scan the phosphor screen using a phosphorimager (Typhoon Trio or equivalent) to produce an image file with a gel extension. See Figure 16 for representative cleavage intensity scans of two lanes from a phosphor image containing deuterated (black) and nondeuterated (red) SRL RNA.
Measurement of the deuterium kinetic isotope effect on hydroxyl radical cleavage
Process the gel file by importing it into SAFA [20] (a computer program for semi-automated footprint analysis). Use SAFA to fit and integrate the gel bands in each of the lanes containing a hydroxyl radical-cleaved RNA sample.
A deuterium kinetic isotope effect is present when the calculated ratio (kH/kD) is greater than 1.0. Residues lacking deuterium should have a kH/kD ratio of 1.0 and so can be used as internal standards to help assess the quality of the data.
CONCLUSIONS
The protocol described here has been used to synthesize specifically deuterated analogs of each ribonucleoside 5′-triphosphate. As an example of the utility of deuterated nucleotides for RNA structural biology, five deuterated analogs of each ribonucleotide were synthesized and incorporated into the SRL RNA molecule to study mechanistic and structural aspects of RNA strand cleavage by the hydroxyl radical [8]. We anticipate that the use of specifically deuterated nucleotides for chemical probe analysis of RNA will be valuable in the structural elucidation of the rapidly increasing collection of RNA molecules that continue to be discovered. This synthesis protocol will contribute to this effort by facilitating the easy availability of specifically deuterated ribonucleotides.